Article Text

Abstract
Mutations within the RIEG1 homeobox gene on chromosome 4q25 have previously been reported in association with Rieger syndrome. We report a 3′ splice site mutation within the 3rd intron of the RIEG1 gene which is associated with unilateral Peters’ anomaly. The mutation is a single base substition of A to T at the invariant -2 site of the 3′ splice site. Peters’ anomaly, which is characterised by ocular anterior segment dysgenesis and central corneal opacification, is distinct from Rieger anomaly. This is the first description of a RIEG1 mutation associated with Peters’ anomaly.
- RIEG1 gene
- Peters’ anomaly
Statistics from Altmetric.com
Peters’ anomaly, a severe form of anterior ocular dysgenesis, is characterised by central corneal opacification secondary to a posterior corneal defect.1 There is usually iris/lens material adherent to the corneal defect. The condition is bilateral in 80% of cases, carries a high risk of early onset glaucoma, and is often associated with a poor visual outcome. Sporadic, autosomal dominant, and autosomal recessive forms have been described.2 3 The aetiology is diverse and the majority of cases have no identifiable cause. A small number of cases have been described in association with PAX6 mutations, while chromosome abnormalities (including aneuploidies, translocations, and deletions) and environmental agents (such as maternal alcohol ingestion) may also be implicated.4-6 Peters’ anomaly is often an isolated finding but may be associated with multiple systemic abnormalities.6 7 In the case of Krause-Kivlin syndrome (Peters’ plus syndrome, MIM 261540), Peters’ anomaly is found, in association with skeletal and cardiac abnormalities and mental retardation, as one feature of a recognisable, multisystemic, autosomal recessive dysmorphic syndrome.8 9
Rieger syndrome is an autosomal dominant condition which is also characterised by anterior segment abnormalities.10 These include posterior embryotoxon, iris stromal hypoplasia, and abnormalities of pupil shape (corectopia) and number (polycoria). There is a 50% risk of glaucoma. A number of systemic associations are described of which the most common are dental abnormalities (anodontia/hypodontia) and umbilical abnormalities. The condition is heterogeneous: a locus on chromosome 4q25 has been identified by genetic mapping following the observation of chromosomal rearrangements of this region in patients with Rieger syndrome.11-13Recently mutations within the RIEG1 gene at 4q25 have been found in patients with Rieger syndrome.14 This homeobox gene encodes a homeo domain characteristic of the bicoid related proteins and consists of four exons. A second locus for Rieger syndrome has been mapped to 13q14.15
The homeobox region of RIEG1 traverses exons 3 and 4. We report a patient with Peters’ anomaly unilaterally and cataract and iris hypoplasia in the contralateral eye who has a 3′ splice site mutation in the third intron of the RIEG1 gene on chromosome 4q25. This is the first report of a RIEG1 mutation in a patient with Peters’ anomaly and suggests that a range of anterior ocular abnormalities might be caused by mutations in this gene.
Case report
The patient was found to have central corneal opacification of the right eye at birth. Pregnancy was normal with no history of intrauterine infection or exposure to teratogenic agents and delivery at term (weight 2680 g) was uncomplicated. A TORCH screen was carried out and was shown to be negative.
Ophthalmic examination was performed under anaesthetic at 4 days. Anterior segment examination showed bilateral abnormalities. In the right eye there was corneal ectasia, with central clouding and peripheral vascularisation consistent with a diagnosis of Peters’ anomaly. There was scleral thinning temporally and no red reflex. In the left eye there was evidence of a milder anterior segment dysgenesis with remnants of a persistent pupillary membrane, areas of iris hypoplasia, and an anterior polar cataract (fig 1). Horizontal corneal diameters were 11 mm (R) and 9.5 mm (L) and intraocular pressures 37 (R) and 14 (L). Fundus examination was only possible in the left eye and showed no abnormality.
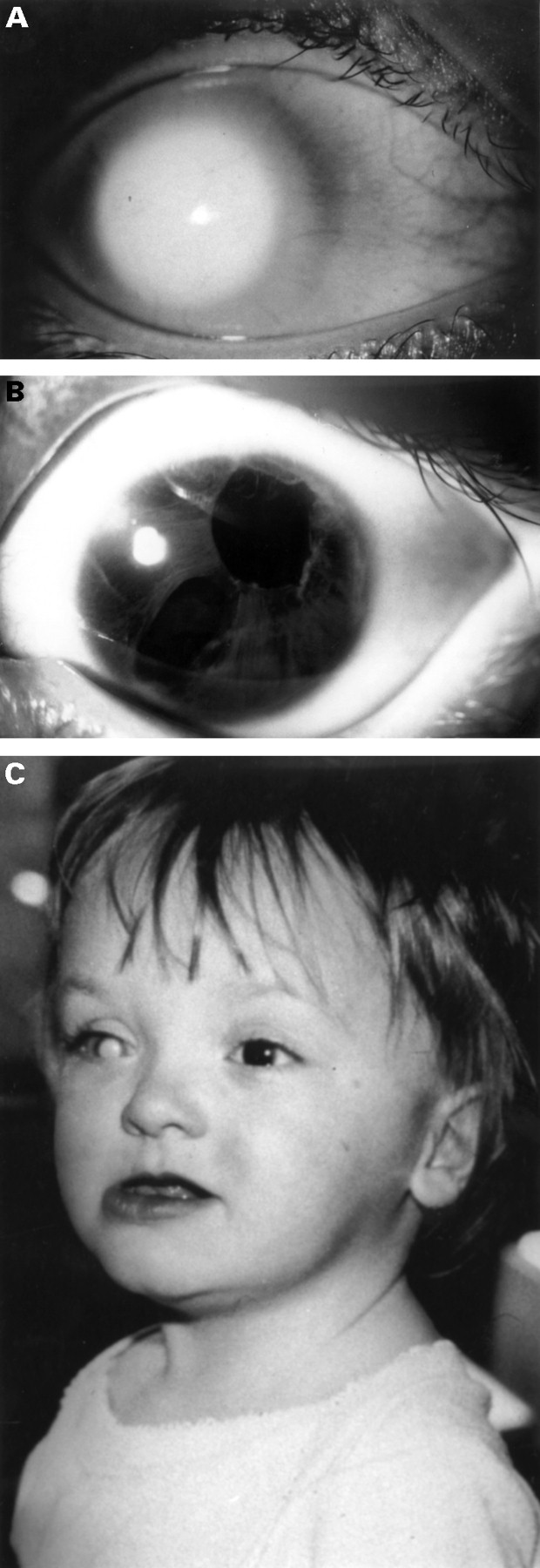
Clinical features of patient. (A) Anterior segment of right eye showing classical Peters’ anomaly. (B) Anterior segment of left eye showing polycoria and iris hypoplasia consistent with Rieger anomaly. (C) Facial features.
Full clinical examination at the age of 18 months showed subtle dysmorphic features, with slightly cupped ears, midface hypoplasia, and mild prognathism. Dental examination showed evidence of hypoplastic teeth with large gaps between them. There was failure of involution of the umbilical skin.
By the age of 4 years, the patient had developed a manifest horizontal nystagmus. Vision in the left eye was 6/18. The appearance in the right eye of unilateral Peters’ anomaly was largely unchanged and the eye was blind. In the left eye there was now evidence of iris hypoplasia with polycoria. On that side, the anterior polar cataract was unchanged and fundal examination showed evidence of foveal hypoplasia. The left intraocular pressure has remained within normal limits. She had significant dental hypoplasia.
The proband’s mother had normal vision and no abnormality was found on examination. The father, with whom the family had no contact, was known to have nystagmus and to have developed glaucoma at the age of 14 years, as had his father. He was also known to have poor dentition.
MOLECULAR ANALYSIS
PCR primers were designed to amplify the coding sequence of exons 3 and 4 of the RIEG1 gene. In the patient, SSCP of the PCR product from the 5′ end of exon 4 showed a mobility shift which was not present in 120 control chromosomes of similar ethnic background (fig 2A). SSCP of the remainder of the coding sequence showed no abnormalities. Direct sequencing of the heterozygous PCR product in both forward and reverse directions showed an A to T substitution at the invariant −2 site of the 3′ splice site of intron 3 (fig 2B). This sequence change was not present in a normal control or in the mother. Paternal samples were not available for analysis.

{kind=link}
{kind=link}
Molecular analysis of patient. (A) SSCP analysis of PCR product from 5′ of exon 4 of the REIG1 gene; the patient (arrowed) shows a mobility shift which is not seen in 25 control samples. (B) Reverse direction sequence analysis of patient (1), patient’s mother (2), and control (3). The patient is heterozygous at position 128. This represents an A to T substitution of the invariant −2 site of the 3′ splice site which is not seen in either her mother or a normal control.
Discussion
Peters’ anomaly represents the morphological description of one form of anterior segment dysgenesis. The characteristic disorganisation of the anterior segment relates to disruption of posterior corneal development and its subsequent opacification and adhesion to the lens/iris. The effects on the anterior segment are heterogeneous and can include many of its structures. Perhaps not surprisingly, therefore, the underlying aetiologies of the condition are diverse and include developmental, environmental, and genetic causes. Of the latter, PAX6 mutations have been described in a small number of patients.4 A small number of families have been described either with phenotypes which overlap between different anterior segment dysgeneses or with segregation of both Peters’ and Rieger anomalies in the same family.2 15 16 Here we report a patient who has a mutation in the RIEG1 homeobox gene on 4q25, the first molecular confirmation that RIEG1 mutations can cause both conditions.
Expression studies during development indicate that the major expression of RIEG1 in the embryonic eye, as assayed by whole mount in situ hybridisation of mouse embryos, is in ocular mesenchyme.14 This includes those structures derived from cells of both primitive mesoderm and neural crest origin. Within the anterior segment, the latter derivatives contribute much of the connective tissues, including the corneal stroma and endothelium, uveal melanocytes, and iris stroma, and the ciliary body structures. Therefore a common pathway may be inferred as the underlying cause of Peters’ anomaly secondary to mutations of both PAX6 and REIG1, an alteration of the development of the derivatives of the neural crest, in particular those structures relating to posterior corneal development. In Peters’ anomaly, the resultant morphology presumably represents one of many potential endpoints of neural crest disruption during embryogenesis rather than a separate entity in itself.
Mutations in the 3′ splice site which are associated with phenotypic effects are well recognised and of these the −2 invariant adenine residue, such as is described here, is the commonest site for such a mutation.17 Of the six RIEG1 mutations originally described associated with Rieger syndrome one was found within intron 3, 11 bp upstream of the 3′ splice site which is thought to create a novel splice acceptor upstream of the correct one. This is therefore the second 3′ splice site mutation to be described within intron 3 which is associated with phenotypic effects.
It is of note that at the age of 18 months our patient had signs of extraocular associations, including mild failure of involution of umbilical skin and subtle signs of abnormal dental development. These phenotypic features may be good clinical indicators of a RIEG1 mutation in patients with anterior segment dysgenesis.18 Families with variant ocular phenotypes, including iridogoniodysgenesis and autosomal dominant iris hypoplasia, have recently been shown to result from mutations in the RIEG1 gene.19 20 Although not present in all patients, extraocular manifestations were present in both conditions. By contrast, it is interesting to note that the family with Rieger syndrome which is linked to chromosome 13q does not have umbilical abnormalities.15
Acknowledgments
RP is supported by the Birth Defects Foundation. GB is a Research Clinician Scientist Fellow supported by the Wellcome Trust (Reference 51390/Z). We are grateful to Jeff Murray and Elena Semina for assistance with primer design.