Article Text

Abstract
Angelman syndrome ( AS) is a neurodevelopmental disorder characterised by severe learning difficulties, ataxia, a seizure disorder with a characteristic EEG, subtle dysmorphic facial features, and a happy, sociable disposition. Most children present with delay in developmental milestones and slowing of head growth during the first year of life. In the majority of cases speech does not develop. Patients with AS have a characteristic behavioural phenotype with jerky movements, frequent and sometimes inappropriate laughter, a love of water, and sleep disorder. The facial features are subtle and include a wide, smiling mouth, prominent chin, and deep set eyes. It is caused by a variety of genetic abnormalities involving the chromosome 15q11-13 region, which is subject to genomic imprinting. These include maternal deletion, paternal uniparental disomy, imprinting defects, and point mutations or small deletions within the UBE3A gene, which lies within this region. UBE3A shows tissue specific imprinting, being expressed exclusively from the maternal allele in brain. The genetic mechanisms identified so far in AS are found in 85-90% of those with the clinical phenotype and all interfere with UBE3A expression.
- Angelman syndrome
- chromosome 15
- genomic imprinting
Statistics from Altmetric.com
In 1965, Harry Angelman, an English paediatrician, reported the clinical findings in three children with similar features of severe learning disability, ataxic, jerky movements, inability to speak, and easily provoked laughter. All three had epileptic seizures with a characteristic EEG appearance and subtle dysmorphic facial features.1 This condition, originally known as the “happy puppet” syndrome, is now known by the less pejorative term of Angelman syndrome. For over 20 years it was considered a rare disorder, and although the occurrence of families with affected sibs suggested a genetic aetiology, no known cause could initially be identified. In 1987 Magenis et al2 identified a deletion of chromosome 15q11-13 in two patients with Angelman syndrome and subsequent work has shown that Angelman syndrome can be caused by a variety of genetic mechanisms which involve this imprinted region of the genome. All of these mechanisms affect maternal expression of the UBE3A gene which lies at the 15q11-13 locus.3 In recent years clearer delineation of the clinical phenotype of Angelman syndrome and improved diagnostic testing has led to improved recognition of the condition and the incidence of Angelman syndrome is now estimated to be between 1 in 10 000 and 1 in 40 000.4–7
Studies of the specific cognitive and behavioural features associated with AS8 and of the seizure disorder have improved management of the condition and provided insight into the long term outlook for affected patients. Molecular genetic studies have begun to elucidate the role of genes within the 15q11-13 region in the pathophysiology of Angelman syndrome9 and have also shed light on the more general phenomenon of genomic imprinting.10 We review the clinical features, natural history, and current management of the condition and summarise the complex genetic mechanisms involved.
CLINICAL FEATURES
The original patients reported by Angelman1 all had severe learning disability, epileptic seizures, ataxia, absent speech, and dysmorphic facial features with a prominent chin, deep set eyes, wide mouth with protruding tongue, and microcephaly with a flat occiput. They were also hypopigmented with fair hair and blue eyes (fig 1). Although many patients with AS have these characteristics,11 it is now clear that the clinical spectrum of Angelman syndrome is much broader than was originally thought. Many patients are normally pigmented and some have a normal head circumference. Seizures are not present in every case and some speech may be present. Some patients may not have the characteristic dysmorphic facies and have minimal ataxia. Several patients with AS are able to speak, although speech is always limited. The behavioural features seen in AS are perhaps the most consistent clinical feature. It is necessary, therefore, to have a wide index of clinical suspicion in a child with the behavioural phenotype of the condition as described below. A summary of the main clinical features and their frequencies is given in table 1.
Main clinical characteristics of AS

(A) AS patient with 15q11-13. (B) AS patient with uniparental disomy. (C) AS patient with imprinting defect. (D) AS patient with UBE3A mutation.
BEHAVIOURAL CHARACTERISTICS
The behavioural characteristics of AS are striking and it is these which often prompt clinicians to consider the diagnosis. They are present in all patients irrespective of the type of genetic abnormality. Paroxysms of easily provoked laughter begin within the first few weeks of life and almost all patients are happy and smile frequently. Laughter is usually provoked, but the stimulus is often minimal and the laughter can be inappropriate. Hyperactivity and sleep disturbance are common in childhood and can pose major management problems. The sleep disorder can be improved by behaviour therapy with adherence to a strict bed time regimen and by the use of melatonin, which is effective in around 50% of patients.12 People with Angelman syndrome love water and have a fascination for reflective surfaces, plastic, and balloons. They enjoy being in the company of others and watching TV, especially slapstick humour. With progression into adulthood the behaviour becomes quieter and concentration span increases. The sociable disposition still persists and paroxysms of laughter may occur. Some adults have shown a tendency to aggressive behaviour, especially if frustrated because of difficulty with communication.
COMMUNICATION AND SELF HELP SKILLS
Communication difficulties are a prominent feature of Angelman syndrome.13 Speech does not develop and most AS patients have a vocabulary of only two or three words.14 Most patients will be able to understand simple commands within the context of their daily routine. A minority can communicate using a formal sign language such as Makaton or the Picture Exchange Communication System (PECS). Others use gestures to communicate. Some patients have been able to use augmented communication devices (for example, Dynavox) to good effect and acquisition of communication skills is often easier as patients get older and concentration span improves. Difficulties with communication can lead to frustration. Speech is present in a few patients (see Phenotype/genotype correlation ), but even these will have difficulty in understanding complex commands and lack capacity for abstract thought.
Self-help skills in Angelman syndrome vary. Most patients learn to walk and to communicate their likes and dislikes. They can make choices, for example, regarding the food they eat and the clothes they wear. Help is needed with dressing and bathing, though undressing is easier. Toilet training is possible and about a third of patients will be dry by day, fewer by night. People with AS can usually feed themselves using basic utensils. All require supervision as they have no sense of danger. Although patients are not able to live independently, many have a good quality of life with a semi-independent existence with round the clock supervision.
NEUROLOGICAL FEATURES OF ANGELMAN SYNDROME
All AS patients have severe mental retardation and delayed motor milestones. They sit unsupported at around 12 months, crawl (commando style) or bottom shuffle at 18-24 months, and walk at a mean age of 4 years (range 18 months to 7 years).5 The gait is slow, stiff legged, and ataxic and the arms are raised and held flexed at the wrists and elbows. Hand flapping is common when walking and if excited. Muscle tone is abnormal with truncal hypotonia and hypertonicity of the limbs and reflexes are brisk. Thoracic scoliosis occurs in approximately 10% of children but is a major problem in the majority of adult patients. It may require orthopaedic treatment by physiotherapy or bracing and although these may be sufficient to resolve the scoliosis in some patients, in other patients the deformity is progressive and surgery is required. Many patients with AS have jerky movements and a discrete tremor of the fingers owing to cortical myoclonus. This phenomenon is worsened by stress and is more pronounced in adults, some of whom develop increasing tremor.
SEIZURES IN ANGELMAN SYNDROME
Epileptic seizures occur in 80% of patients. Age at onset varies between one and five years. The initial presentation of the epilepsy is with febrile convulsions in infancy.15,16 In childhood a variety of seizures can be observed, ranging from tonic-clonic seizures, atypical absences, complex partial, myoclonic, atonic, and tonic seizures to status epilepticus. Absence status and myoclonic status may also occur. Earlier reports suggested a decreasing frequency of epileptic seizures with age,5,17,18 but further follow up has shown that although there may be a relatively quiescent period during late childhood and adolescence, a large number of adults have epileptic seizures, particularly atypical absences or myoclonic seizures.19 The epilepsy of Angelman syndrome is difficult to control with antiepileptic drugs, especially in childhood. The most effective drugs are sodium valproate as monotherapy or in combination with clonazepam or other benzodiazepines. Carbamazepine sometimes has an adverse effect.19,20 In adults, phenobarbitone is also effective. Experience with the newer antiepileptic drugs is very limited.
EEG FINDINGS IN ANGELMAN SYNDROME
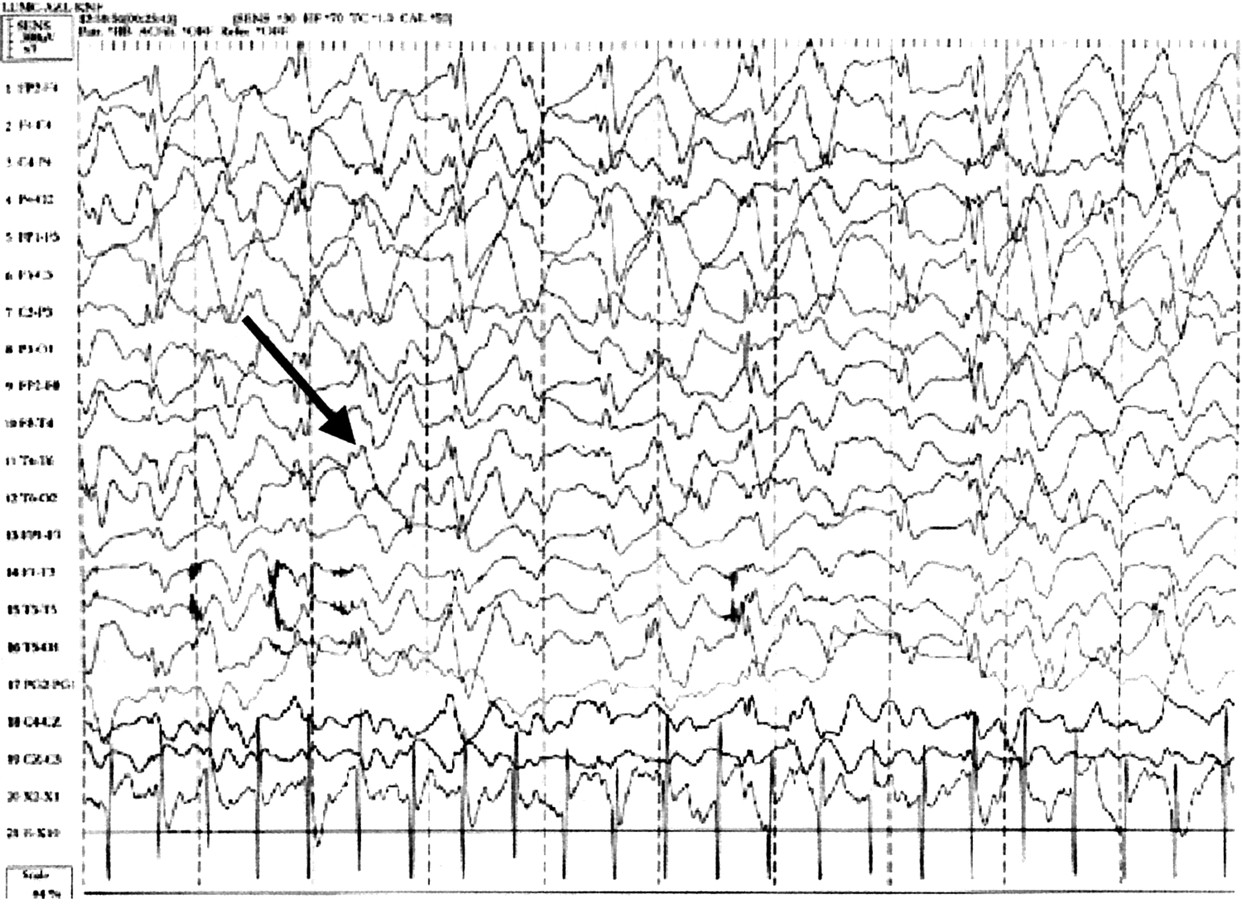
There are specific EEG patterns in AS patients which appear in isolation or in different combinations. They are similar in patients both with and without seizures.20,21 In childhood the three characteristic patterns are (1) persistent rhythmic 4-6/s activity reaching more than 200 μV, not associated with drowsiness, (2) prolonged runs of rhythmic (triphasic) 2-3/s activity with an amplitude of 200-500 μV, maximal over the frontal regions and normally mixed with spikes or sharp waves, and (3) spikes mixed with 3-4/s components, usually of more than 200 μV mainly posteriorly and facilitated by or only seen on eye closure. In patients over 10 years of age the background rhythm is slower than normal. There may be focal spikes and intermittent or continuous triphasic delta activity (fig 2), maximal over the frontal regions.15 There is no correlation between the EEG findings and the paroxysms of laughter seen in AS. The EEG features are specific to AS and are an important diagnostic feature. Unfortunately, these three components may not always be seen within the same EEG recording and may occur at different times in the same patient.15,18,22 It is therefore worth repeating the EEG if the diagnosis of AS is strongly suspected but the typical features are not seen initially.

Characteristic Angelman EEG. Triphasic delta wave activity arrowed.
ANGELMAN SYNDROME IN ADULTS
The phenotype of Angelman syndrome is an evolving one which changes with progression into adulthood (fig 3). Puberty occurs at a normal time and there are normal secondary sexual characteristics. Facial characteristics in adults are more pronounced with marked mandibular prognathism, pointed chin, macrostomia, and a prominent lower lip.17,19,23,24 Eyes appear more deeply set and in some patients keratoconus (because of eye rubbing) is observed. Mobility decreases because of limb hypertonicity, development of thoracic scoliosis, and a general reluctance to walk.19 Some patients develop contractures and in those with reduced mobility ataxia is less obvious. Many patients become obese and oesophageal reflux may occur and can be severe. Despite these problems a good quality of life is maintained into adulthood and in the majority of patients lifespan is not greatly reduced.

Facial features of Angelman syndrome in a young adult.
GENETICS OF ANGELMAN SYNDROME
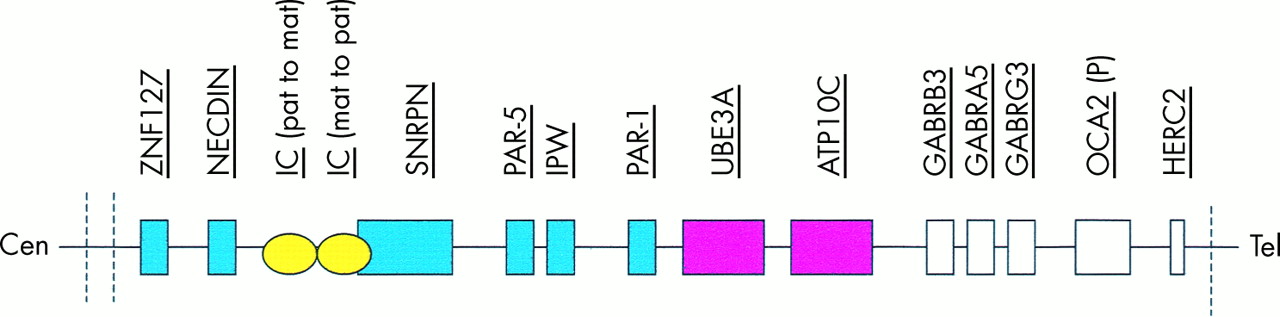
There are four major genetic mechanisms which are now known to cause Angelman syndrome and AS patients have been divided into classes I to IV based on these mechanisms.3 A further group of patients, designated class V, have clinical features of AS but no demonstrable cytogenetic or molecular abnormality of chromosome 15q11-13. A summary of the various genetic mechanisms and their frequencies is shown in table 2 and a diagrammatic representation of the 15q11-13 region is shown in fig 4.
Genetic mechanisms giving rise to Angelman syndrome

The Angelman/Prader-Willi region on chromosome 15q11-13 which spans 4 Mb. The common breakpoints are represented by dashed lines. Paternally imprinted genes are depicted in blue and maternally imprinted genes in pink. Those depicted in white are not known to be imprinted. The imprinting centre (IC) is bipartite. The centromeric portion is responsible for the paternal to maternal imprint switch and deletions in this part cause AS. For further details see text.
The commonest genetic mechanism giving rise to Angelman syndrome, occurring in approximately 70-75% of patients, is an interstitial deletion of chromosome 15q11-13. The majority of deletions are of a similar size, approximately 4 Mb, and with common breakpoints. They are thought to occur because of unequal crossing over between low copy repeats (duplicons) within the 15q11-13 region.25 The duplicons contain the transcript of the HERC2 gene, a highly conserved gene which, when mutated in mice, affects development and fertility.26 Most deletions occur de novo and are of maternal origin, in contrast to the 15q11-13 deletions observed in Prader-Willi syndrome which are of paternal origin.27 The common deletion can be detected by FISH analysis and by methylation analysis of the SNRPN (small nuclear ribonucleoprotein polypeptide N) promoter which lies within a CpG island at 15q11-13. In the presence of a maternal deletion only the paternal, unmethylated pattern will be detectable.28 Rarely, patients with AS will have a different sized deletion, often in association with an unbalanced translocation or chromosome rearrangement.29,30 It is important to distinguish this group of patients as the mother may carry the same chromosome rearrangement and therefore be at risk of having a further child with AS. Several patients have been reported in whom a supernumerary inv dup(15) was present in addition to a de novo deletion of 15q11-13.31 It has been hypothesised that the presence of such a marker predisposes to 15q11-13 deletion. Cytogenetic analysis and FISH analysis to exclude cryptic translocation should therefore be carried out on people with AS where the families concerned have requested genetic counselling.
A second class of AS patients, class II, have uniparental disomy (UPD) for chromosome 15 and thereby fail to inherit a maternal copy of UBE3A. The UPD is usually for the entire chromosome and Robinson et al32 have studied in detail the mechanisms by which it arises, concluding that in most cases the error is postzygotic, although some cases have been shown to arise through meiotic non-disjunction. Maternal and paternal ages of patients with UPD are significantly higher than the general population.33 This is likely to be responsible for a higher rate of non-disjunction in patients where UPD has arisen because of a meiotic error, but the mechanism by which raised paternal age contributes to a postzygotic duplication of chromosome 15 is unclear. In some cases UPD(15) arises in association with a Robertsonian translocation or a reciprocal translocation involving chromosome 15.34,35 The occurrence of uniparental disomy is sporadic and accounts for only 2-3% of cases of AS.
Class III patients are those without deletions or UPD, but with abnormal chromosome 15 methylation, signifying a defect in imprinting. Imprinting defects account for 3-5% of patients with AS. This is the process by which epigenetic marking of the chromosomes occurs in the germline, such that they retain a memory of their parental origin.10 Buiting et al36 identified an imprinting centre within chromosome 15q11-13 which regulates chromatin structure, DNA methylation, and gene expression through cis acting elements. This imprinting centre (IC) has a bipartite structure. One part appears to be involved in switching a paternal imprint to a maternal imprint during gametogenesis, whereas the other part is responsible for the paternal to maternal switch. Around 50% of subjects with imprinting defects have an identifiable mutation within the imprinting centre, but in the remaining patients no mutation can be identified. It is thought that in this latter group imprinting defects are caused by spontaneous pre- or postzygotic events.37 A recent report by Cox et al38 has suggested that intracytoplasmic sperm injection may be a mechanism which interferes with establishment of the maternal imprint in an oocyte and might therefore predispose to Angelman syndrome. Among the group of patients where imprinting centre deletions have been detected, there are several familial cases. Where mutations are not identified, AS is usually a sporadic occurrence. An exception is a family where affected subjects and the unaffected mothers had a submicroscopic chromosome inversion within the imprinting centre.39 For parents of children with imprinting defects who wish to pursue prenatal testing during future pregnancies, Glenn et al40 showed that methylation analysis at the SNRPN locus of DNA extracted from chorionic villus or amniocytes will give a reliable result. It has also been postulated that some patients with atypical clinical features may be mosaic for an imprinting centre defect.41 This group of patients often present with hypotonia and obesity and in some cases it is Prader-Willi syndrome and not Angelman syndrome which is initially suspected.
Class IV patients are those who have been shown to have mutations within the gene encoding ubiquitin protein ligase, UBE3A.42,43 Mutations can be identified in 20% of sporadic patients with normal methylation and in around 75% of familial patients.44,45 The UBE3A gene is imprinted in brain and encodes a ubiquitin protein ligase which is thought to play a role in ubiquitination of proteins within the brain, a process which marks those proteins destined for degradation.46 It is expressed predominantly within the hippocampus and Purkinje cells of the cerebellum and shows tissue specific imprinting, being expressed only from the maternal allele in brain but with biallelic expression elsewhere.47 Mice deficient in maternal Ube3a have impairment of motor skills and spatial learning and exhibit abnormal EEG readings from the hippocampal area.48 There are several substrates for UBE3A, but those which play a critical role in the pathophysiology of AS remain to be identified.9 The UBE3A gene comprises 16 exons with a coding region from exons 8-16. Point mutations have been found throughout the entire coding region with clusters in exons 9 and 16, the latter of which contains a highly conserved HECT domain. The catalytic cleft between the two lobes of the HECT domain is the site of many of the reported mutations.49 Screening of the coding region by SSCP followed by direct sequencing is a relatively reliable way of detecting mutations. Frameshift, nonsense, and splice site mutations have been identified. Some missense mutations have also been reported. Although it is difficult to be certain as to the pathogenicity of these latter changes, one must have a high degree of suspicion where these have occurred de novo or have been inherited from the mother. Several common polymorphisms have also been identified within the UBE3A gene. Where UBE3A mutations have been identified, parental DNA can be analysed to determine if the mother carries the same mutation. The majority of mutations appear to occur de novo and our own experience suggests that only around 20% of mothers will carry the same mutation. There are, however, several reported families where mothers have had more than one affected child despite having had negative UBE3A analysis and these mothers are likely to be gonadal mosaics. For this reason, all mothers of children with UBE3A mutations should be offered prenatal testing in future pregnancies. With the exception of a few mutations which have been found in more than one patient, most are unique. Finally, although rare, Bürger et al50 reported a patient with a 570 bp deletion of UBE3A which was familial and was detected through allelic loss at microsatellite loci. This type of abnormality would not have been detectable on UBE3A sequencing.
There remain some patients with a clinical phenotype of AS where no chromosome 15 abnormality has been identified and these are designated class V patients. Although some would argue that these patients must have alternative diagnoses, our own experience, and that of other groups with significant clinical experience of AS, suggests that class V patients do exist.8 This group is, however, likely to be heterogeneous and to include patients with other disorders.51–58 Some of the diagnoses which must be excluded in this group are Rett syndrome,59,60 the α thalassaemia-mental retardation syndrome,61 and the condition described by Mowat et al, which is now known to be the result of large scale deletions or point mutations within the ZFHX1B gene on chromosome 2.62,63 The diagnosis of AS may also be considered in children with broader symptom complexes, such as cerebral palsy, Lennox-Gastaut syndrome, or mitochondrial disorders.15,22,64 Williams et al65 summarised many of the conditions which mimic Angelman syndrome and a comprehensive list is given in table 3.
AS: differential diagnosis
Other explanations for the existence of this group include the possibility of a UBE3A mutation within a non-coding region or a mutation within another gene in the ubiquitin pathway which affects UBE3A expression. No mutations have been identified to date within the 3′ prime region of UBE3A and there are no reports so far documenting expression of UBE3A within the brain of class V patients. There is an antisense transcript of UBE3A66 but this is paternally expressed and mutations within this gene are not known to be involved in the pathogenesis of AS. One possible candidate, however, is the ATP10C gene67 which is located within 200 kb of UBE3A and has been shown to be maternally imprinted in brain. To date, no mutations have been identified within ATP10C in humans.
GENETIC COUNSELLING
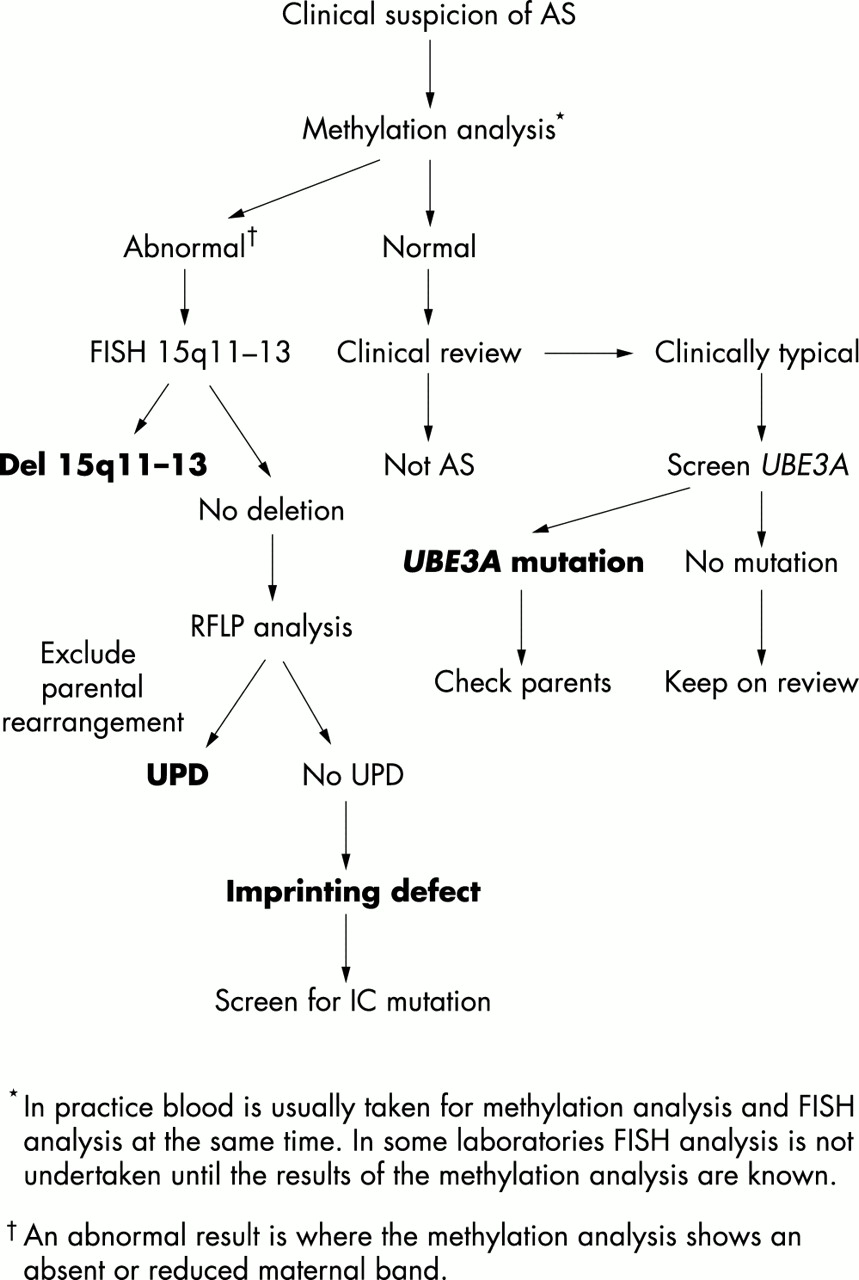
Genetic counselling in AS families is complicated in view of the wide variety of genetic mechanisms which can all give rise to this condition. It is recommended that diagnostic investigation should begin with 15q11-13 methylation analysis, which is relatively inexpensive and will be abnormal in the majority of cases of AS. One can then move on to other tests to elucidate the specific genetic mechanism involved or to screen for UBE3A mutations in patients with suggestive clinical features, EEG, and normal methylation. An algorithm for genetic testing is shown in fig 5. Recurrence risks in AS will depend on the genetic mechanism involved and on whether the mother is shown to carry a genetic abnormality of 15q11-13. For patients with de novo deletions of 15q11-13 and uniparental disomy, recurrence risks are low, although there has been one reported incidence of recurrence owing to possible gonadal mosaicism.68,69 Where imprinting centre mutations and UBE3A mutations are concerned, the situation is complicated further by the high levels of gonadal mosaicism which appear to be present in the mothers.70,71 In these situations, even if the mother tests negative for a mutation identified in a child there is still an appreciable risk of recurrence owing to gonadal mosaicism and prenatal testing should be offered. If the mother carries the same mutation, the recurrence risk is 50%. Recurrence has been reported within families of class V patients and genetic counselling needs to reflect this. This group is likely to include some patients with X linked or autosomal recessive disorders. In some families prenatal diagnosis has been offered by haplotyping the fetus during a future pregnancy to determine whether the fetus has inherited the same maternal chromosome 15 as the affected child. However, this can only be offered in families where the clinical diagnosis of AS is very secure and parents should be made fully aware of the limitations of this type of testing.69,72

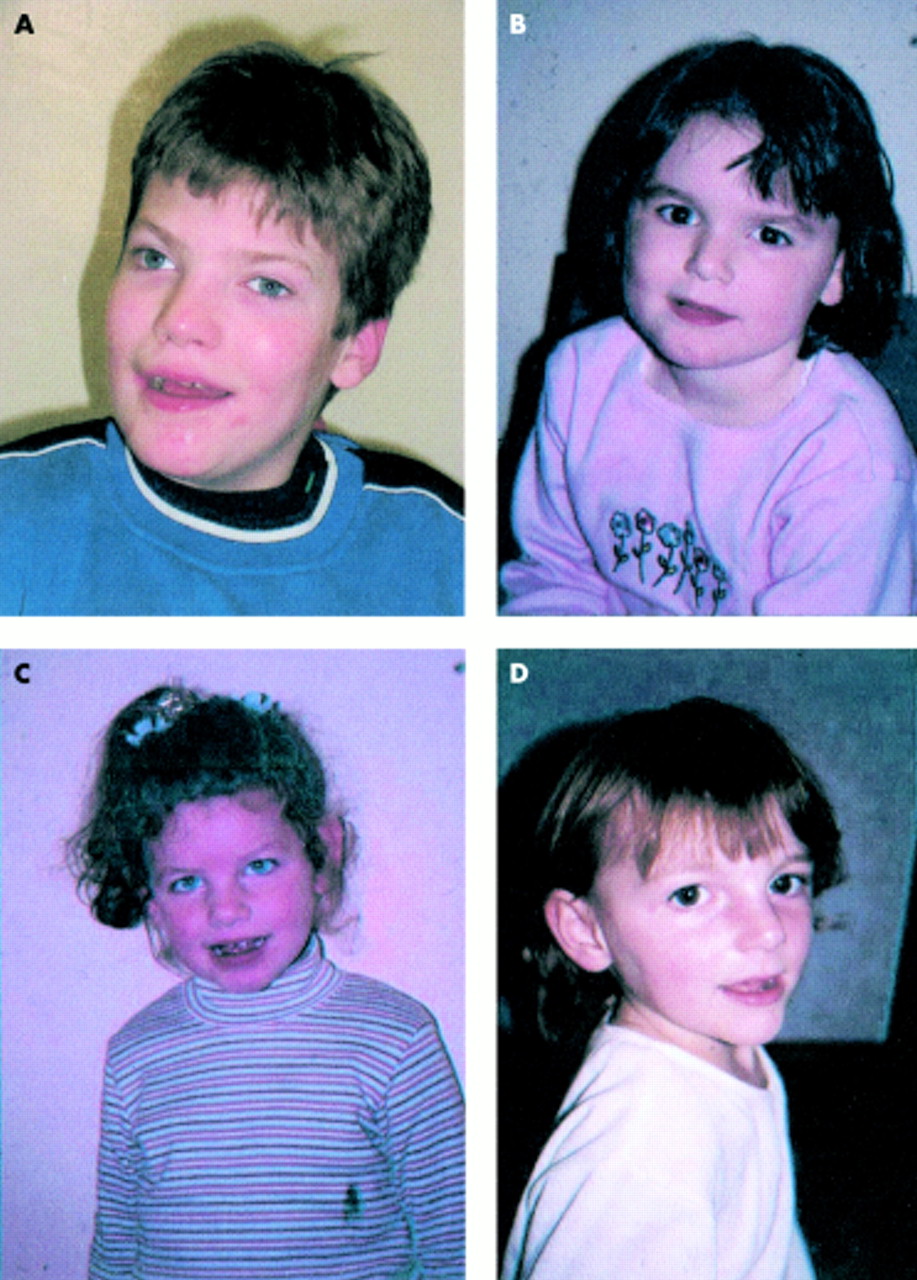{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple algorithm for genetic testing in Angelman syndrome.
PHENOTYPE/GENOTYPE CORRELATION
Delineation of the different genetic mechanisms giving rise to AS has helped to explain some of the phenotypic differences between patient groups (fig 1).8,41,72–76 It has been shown, for example, that patients with chromosome 15 deletions are in general the most severely affected. They have a higher incidence of seizures, microcephaly, and hypopigmentation, greater delay in motor milestones, and absent speech. The more severe features are thought to be the result of haploinsufficiency for a number of genes within the 15q11-13 region. Minassian et al20 suggested that the severe epilepsy seen in AS patients with deletions is the result of the absence of one copy of the GABA receptor genes. The locus for the P gene which is implicated in type II oculocutaneous albinism also lies within this region.77 Recent studies have shown that AS deletion patients with hypopigmentation frequently have a mutation of the P gene on the remaining chromosomal homologue78,79 and some patients have the characteristic ocular findings of albinism with misrouting of optic nerve fibres at the optic chiasm.80 Patients with uniparental disomy of chromosome 15 have a low incidence of seizures, microcephaly, and hypopigmentation. Many of these patients are able to say a few words and their growth parameters are also greater than the deletion group, often being in the upper half of the normal range. Dysmorphic features are less obvious in this group, although the behavioural characteristics are quite typical. AS subjects with imprinting defects are less likely to have microcephaly, hypopigmentation, or seizures and, again, they are more able than the deletion group with less delay in motor milestones and better communication skills. Growth is better than in deletion patients and obesity is relatively common within this group. The abilities of patients with UBE3A mutations fall somewhere in between those of the deletion group and the UPD group. They frequently have seizures and microcephaly but are not hypopigmented and have better motor and communication skills than the deletion group. Lossie et al8 pointed out that this group have a particularly high frequency of obesity as they get older.
Finally, milder AS phenotypes have been reported in patients with incomplete imprinting defects or mosaicism. Gillessen-Kaesbach et al41 described the clinical phenotype in seven AS patients who presented initially with features of the Prader-Willi syndrome but were subsequently shown to have a chromosome 15 methylation pattern in keeping with AS. These patients presented with hypotonia, obesity, and a milder degree of mental retardation. Methylation analysis in some of these patients showed an atypical pattern with only a faint maternal band. Tekin et al81 have also reported a patient with milder clinical features of AS who was proven to have mosaicism for a 15q11-13 deletion, which was detectable on fluorescence in situ hybridisation but where methylation analysis had proven normal.
MANAGEMENT OF ANGELMAN SYNDROME
The management of AS revolves around appropriate therapies for the physical and neurological problems encountered in this condition and provision for special educational needs, given the very specific cognitive profiles and behavioural features of the condition. In some cases AS patients have undergone courses of intensive therapies similar to the conductive education which has been carried out in many children with cerebral palsy. While some children undergoing this type of treatment have shown short term improvement, for example, in mobility and communication,82 there are no data as yet to suggest that this will offer long term benefit in Angelman syndrome. There is evidence from parents, albeit anecdotal, that massage and aromatherapy can improve hyperactivity and concentration. The UK study suggested that AS people appear to need continuous reinforcement of their skills if they are not to lose them. Treatment of the epilepsy in AS is often difficult, especially in the early years,15,83 and the advice of a paediatric neurologist should be sought. Seizures can take many forms and taking a short video clip of suspected seizures to show to the neurologist is helpful. AS children have global developmental problems, but the most marked problem is with acquisition of language. No single communication method works best in AS so every attempt should be made to find a communication system which works for an individual AS child. There remain some children who have very limited communication skills however much input they receive from parents and therapists. AS children have relatively good social skills and fit in well with others within their peer group. Their inherent inquisitiveness and childhood hyperactivity can often pose management problems, and sleep disorder is one of the most significant issues for parents of young children. Many of the problem behaviours associated with the condition can be improved by a consistent approach, with help from a behavioural therapist if necessary. In adults with recent onset of behavioural problems the possibility of oesophageal reflux should be considered .
ANGELMAN SYNDROME RESOURCES
A large amount of knowledge has been gathered over the last 10 years about the clinical features, natural history, and genetic mechanisms involved in Angelman syndrome. Studies of the condition have also been carried out by a variety of professionals involved in the management of patients with AS. Many countries worldwide now have support groups for those with AS and their families and these groups have produced a large amount of information on different aspects of the condition and have in many cases been instrumental in bringing together parents and professionals involved in the care of AS people. An international umbrella organisation now exists (www.iaso.com) and can guide parents and professionals to a variety of AS resources and information about the individual national AS groups. ASSERT, the UK support group, has a free telephone line for parents and a professional helpline.