Article Text

Abstract
Background Split hand and foot malformation (SHFM) refers to a genetically heterogeneous developmental disorder of the hands and feet that presents as median ray deficiency of varying severity. 7q21.3 (SHFM1) is one of six loci described to date, and although DLX5 and DLX6 are compelling candidates in that locus, no intragenic mutations have been described in either of these genes.
Methods The authors combined autozygome analysis and exome sequencing to study a consanguineous family with a highly unusual SHFM phenotype, where there is associated dorsalisation of the hands.
Results A novel missense mutation in a highly conserved residue of the homeobox domain of DLX5 was identified. Unlike previously reported position effect mutations in SHFM1, this first documented intragenic DLX5 mutation is also accompanied by abnormal dorsal-ventral patterning.
Conclusion This study identified the first intragenic DLX5 mutation in SHFM and raises interesting possibilities about a dual role for DLX5 in limb development.
- SHFM1
- DLX5
- dorsalisation of the palm
- palmar nail
- congenital heart disease
- developmental, genetics
- metabolic disorders
- neurosciences
- metabolic disorders
- gene therapy
- molecular genetics
- clinical genetics
Statistics from Altmetric.com
- SHFM1
- DLX5
- dorsalisation of the palm
- palmar nail
- congenital heart disease
- developmental, genetics
- metabolic disorders
- neurosciences
- metabolic disorders
- gene therapy
- molecular genetics
- clinical genetics
Introduction
Split hand and foot malformation (SHFM) is a term used to describe a distinct developmental defect of the hands and feet in which there is deficiency of the central ray digits, giving rise to claw-hand and claw-foot appearance in severe cases, although the level of severity varies even between the limbs of the same patient.1 Many syndromes are associated with SHFM, but non-syndromic SHFM has long been recognised as a distinct monogenic trait with six loci, designated SHFM1-6, identified to date.2–8 Of the four autosomal dominant loci, only one has been solved at the gene level when it was found that TP63—which is known to be linked to a number of other developmental syndromes such as ADULT syndrome, ectrodactyly, ectodermal dysplasia and cleft lip/palate syndrome, Hay–Wells syndrome, limb–mammary syndrome and Rapp–Hodgkin syndrome—is mutated in SHFM4.5 9 Later, WNT10B mutations were found to underlie SHFM6, the only autosomal recessive SHFM locus.7 The underlying genes in the remaining three dominant and the only X-linked SHFM loci remain elusive.
The first described SHFM locus SHFM1 is particularly interesting because it has been involved in numerous chromosomal aberrations including deletions, inversions and translocations, and yet no single gene has been implicated.10–24 The critical interval has been narrowed by breakpoint mapping of the above reports to a region of 1.5 Mb that encompasses DSS1 as well as two homeobox genes that are arranged tail to tail: DLX5 and DLX6.23 25 Despite the compelling nature of DLX5 and DLX6 as candidate genes based on the Dlx5/Dlx6 double knockout mouse phenotype, no intragenic mutations have been reported in either of these two genes to date.1 26 27
In this study, we conducted positional mapping of a consanguineous family that defined a previously unreported autosomal recessive SHFM1 which was accompanied by remarkable dorsalisation defect of the hands and identified a novel intragenic mutation in DLX5 as the likely cause.
Methods
Patients
Patients and relatives were recruited after they signed a written informed consent form approved by KFSHRC IRB (RAC 2080006). Patients and carrier parents received full clinical genetics evaluation. Venous blood (5–10 ml) was collected in EDTA tubes for DNA studies.
Autozygome analysis
Identification of the full set of genomic regions that are identical by descent per individual (autozygome) was performed as described before.28 29 Briefly, we used Affymetrix Axiom SNP Chip (>560 000single nucleotide polymorphism (SNPs)) for genotyping using the manufacturer's protocol. The resulting genotypes were evaluated for runs of homozygosity using autoSNPa. Linkage analysis was performed on easyLINKAGE using fully penetrant autosomal recessive model with allele frequency of 0.001.
Exome analysis
A DNA sample from the index patient was prepared as an Illumina sequencing library, and in the second step, the library was enriched for the desired target using the Illumina Exome Enrichment protocol. The captured library was sequenced using Illumina HiSeq 2000 Sequencer.
Results
Clinical report
The index patient is 6-year-old Yemeni girl. Her mother's pregnancy was uneventful. Severe limb defects were first noted at birth. Other than delayed walking caused by her lower limb defects, her development has been normal and she uses a wheelchair for mobility. She has a history of hearing impairment but wears no hearing aids. Her parents are healthy first cousins and they have two affected children and several other affected relatives (figure 1). One affected sister passed away at 7 years of age but the cause of death was unknown.

Pedigree of a family with recessive split hand and foot malformation (SHFM). Index patient indicated by arrow.
Physical examination revealed she had mild synophrys and a low anterior hair line. Remarkably, there was near full dorsalisation of the palms, tapered fingers and circumferential (cylinder shaped) nails, as well as asymmetric short and severely deformed legs and feet (figure 2). There was restriction of joint flexion at all the metacarpophalangeal and interphalangeal joints as well as mild scoliosis. Anthropometric measurements revealed severe short stature but normal weight and head circumference. The rest of the physical examination was normal.
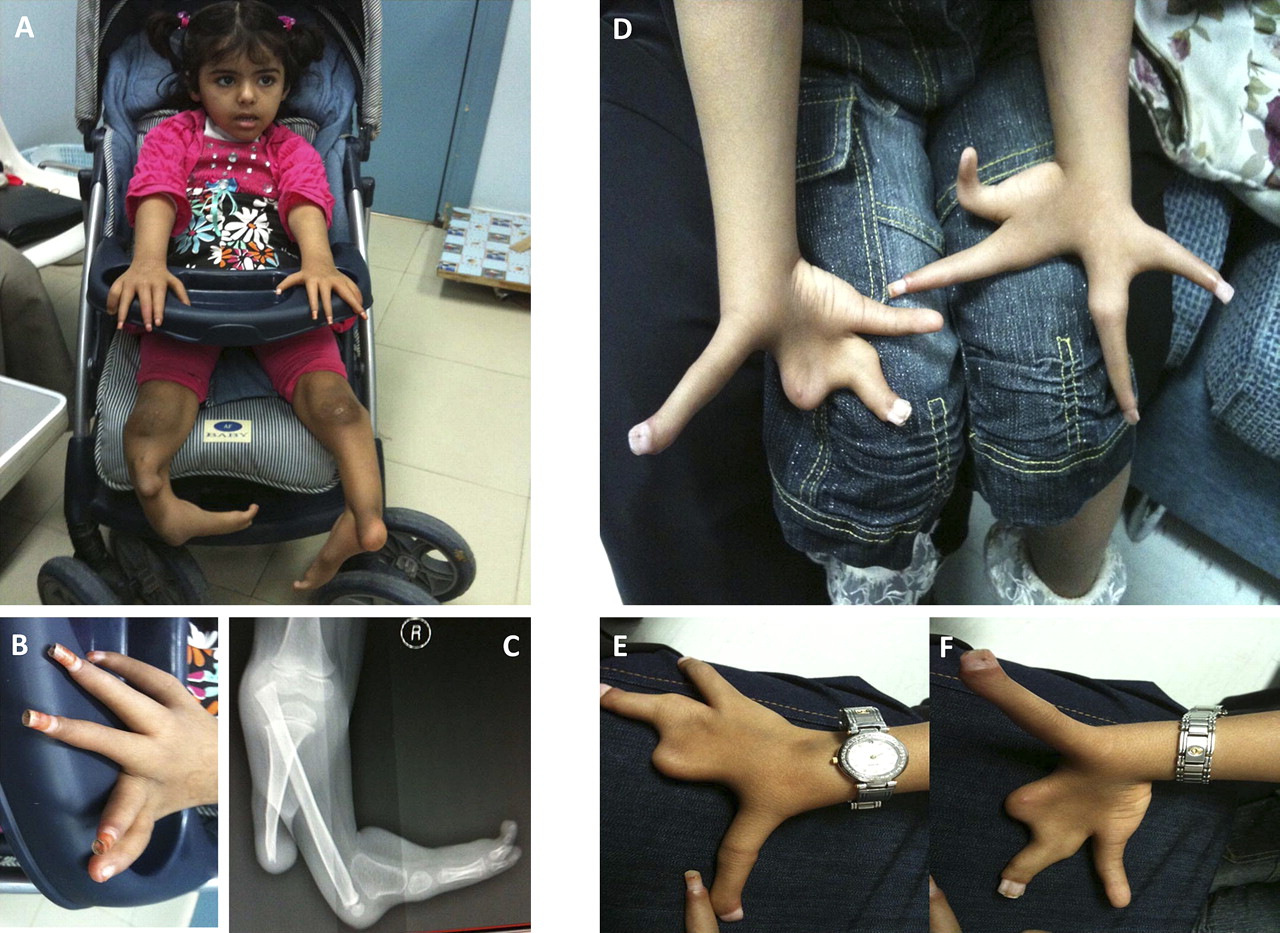
Clinical photographs of the index patient (A). Note the circumferential finger nails shown in panel B and severe lower limb malformation shown on x-ray in panel C. (D) Severe ectrodactyly of the hands of the sister. The complete palmar dorsalisation is clearly shown in panels E and F.
The second affected sister is currently 4 years old and also suffered from hearing loss. She had mild frontal bossing and high frontal hair line. Furthermore, her hands showed a severe asymmetric claw-like deformity. Her right hand had an elongated abducted thumb, wide fused ring and middle fingers, and an absent index finger. In comparison, the left hand had a deep middle cleft, elongated ring finger, and clinodactyly of the fifth finger (figure 2). Her back and lower limbs looked normal.
The following investigations for the index patient revealed normal results: blood karyotype (regular and molecular), abdominal ultrasound, and echocardiography. Audiometric assessment showed moderate hearing impairment in the right ear and severe impairment in the left. A skeletal survey showed hypoplasia of the lower third of the right tibia with rotation of the fibula, malpositioned left tibia and fibula, medially placed distal end of the fibula, subluxation of the superior tibiofibular and ankle joints, and a deviated fifth distal phalanx. A CT angiogram of the lower limbs showed no obvious vascular malformation and free flow of the contrast towards the feet.
Positional mapping
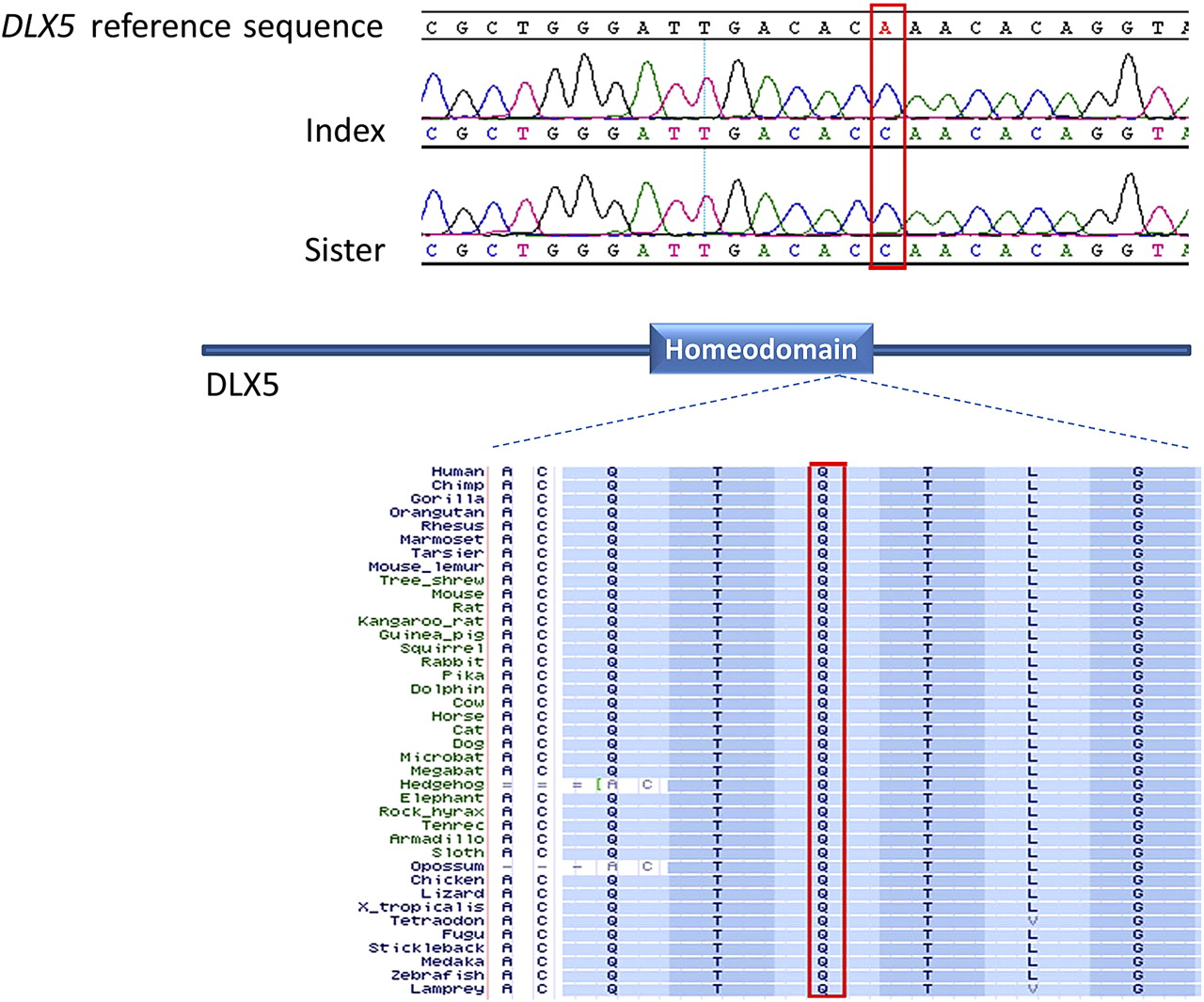
Consistent with the consanguineous nature of the parents, several runs of homozygosity were identified in the index patient and her sister and there was an overlapping pattern on five chromosomes as evident on both autoSNPa and linkage analysis (figure 3). We used this autozygome overlap to filter the >85 000 variants generated on exome sequencing as described previously.29 30 By applying this filter, we were able to narrow the search from 1246 novel variants to just 12 variants present in the shared autozygome of the two sisters. Of these, six variants were present in other Saudi exomes and were thus excluded. Of the remaining six variants, only the missense mutation in DLX5 (NM_005221:exon2:c.A533C:p.Q178P) was predicted to be pathogenic on SIFT and PolyPhen (supplemental table S1). This mutation segregated with the phenotype in the family, affects an amino acid residue that is fully conserved across all species examined, had the highest possible PolyPhen pathogenicity score (0.999) and the lowest tolerance score on SIFT (0), and was not identified in 192 ethnically matched controls or the Exome Variant Server.

Autozygome analysis with runs of homozygosity (ROH) shared between the two siblings shown as blue bars. Below (left) is the result of linkage analysis with peaks corresponding to these ROH, and the 7q locus (right) is shown as black blocks (boxed in red). The 7q locus is flanked by rs11975422 and rs1227192 (Chr7: 90 915 282–112 394 361).
Discussion
SHFM is thought to represent a failure in the maintenance, rather than the formation, of the apical ectodermal ridge (AER) specifically in the median plane.1 Many genes are known to be expressed in this region of the developing limb including the two known SHFM genes TP63 and WNT10B.31 32 DLX5 is one of at least six vertebrate DLX genes that are related to the Distal-less gene of Drosophila. DLX5 and DLX6 are in close proximity to each other on 7q and expression of both in the AER under the control of P63 has been demonstrated.33 34 Dual, but not singular, deficiency of both proteins in mouse has been shown to phenocopy the SHFM phenotype in humans.27 Despite the relatively frequent observation of chromosomal aberrations adjacent to DLX5 and DLX6 in the setting of SHFM, no mutation has been identified in either of these two genes and even the smallest deletion overlap encompassed these two genes in addition to DSS1. This led to speculation that these genomic rearrangements exert a position effect on DLX5 and/or DLX6. In other words, SHFM1 is caused normally by the simultaneous inactivation of DLX5 and DLX6, either by deletions encompassing the genes or by deletions and translocations that disrupt their co-regulation. However, aberrant transcription of these two genes has not been demonstrated in any of those SHFM1 cases.
The family described here offered a unique opportunity to solve the dilemma of the actual disease gene in SHFM1 because affected patients linked to SHFM1 but lacked any detectable chromosomal aberration, making it likely that the disease is caused by a point mutation. The combination of exome/autozygome analysis was possible in this family because of its consanguineous nature and the recessive pattern of inheritance as described before.29 30 Indeed, the critical region on 7q contained one identifiable coding mutation in DLX5 and several lines of evidence support the notion that this mutation is disease-causing. First, DLX5 has long been proposed to be the candidate gene for SHFM1 by positional mapping and by inference from the mouse model (see above). Second, the other variants we identified were predicted to be tolerated in silico whereas DLX5 mutation had the highest pathogenicity score. Third, this mutation affects an absolutely conserved amino acid residue in the homeobox domain of all DLX genes (figures 4 and S1). In fact, missense mutations in the corresponding homeodomain of DLX3 have also been shown to be pathogenic and result in tricho-dento-osseous syndrome.35
{kind=link}
{kind=link}
{kind=link}
{kind=link}
DLX5 sequence chromatogram showing the homozygous mutation NM_005221:exon2:c.A533C:p.Q178P in both patients. The strong conservation of the Q178 residue across species is shown below a schematic of DLX5 where the DNA binding domain (homeodomain, residues 137–196) is highlighted.
The abnormal dorsalisation of the palms is an extremely interesting aspect of the phenotype in this family because it may suggest a dual function for DLX5 in limb patterning. In addition to its critical role in limb outgrowth, AER is also essential for the establishment of the proper polarity that demarcates the ventral and dorsal compartments of the developing limb, a role believed to be mediated largely by BMP4 through its BMP1A receptor.36 When this receptor is inactivated, the delicate balance between the strictly ventral EN1 and strictly dorsal WNT7A is tipped in favour of WNT7A whose expression domain extends as a result to the ventral aspect, leading to a bi-dorsal limb.37 Importantly, it has been suggested that DLX5 acts as a downstream effector of BMP4 induced ventralisation.38–40 Therefore, it might be reasonable to hypothesise that the point mutation we identified in DLX5 affects its role not only in AER controlled limb outgrowth but also in mediating or regulating the BMP induced dorsal–ventral patterning. The interaction between DLX5 and BMP4 is also well documented in the inner ear, which is abnormal in both of our patients.41 There are precedents for differences in phenotype between intragenic and enhancer mutations affecting the same gene. For example, enhancer mutations of SOX9 lead to the Pierre–Robin sequence whereas intragenic mutations are known to cause a lethal form of skeletal dysplasia.42 However, we are not aware of examples where the pattern of inheritance between the two classes of mutation is different. We posit that this is yet another example of the opportunity provided by the study of consanguineous populations in delineating novel mutational mechanisms for diseases that have classically been described as autosomal dominant.43
We cannot exclude the possibility that the phenotype in this family was caused by a non-coding mutation in linkage disequilibrium with the mutation we report. Unequivocal proof that Q178P is a bona fide disease-causing mutation can come from the identification of the same mutation in a family with the same phenotype but on a different haplotype or from the generation of an animal model. However, the latter may not be straightforward given the interspecies differences in the functional annotation of this locus, some of which have already been mentioned above—for example, only homozygosity for Dlx5/Dlx6 double knockout can recapitulate the dominant phenotype observed for the SHFM1 locus.
In summary, this report describes the identification of the first intragenic DLX5 mutation in a family with an unusual SHFM that is recessively linked to SHFM1 and is associated with defects in both limb outgrowth as well as dorsal–ventral patterning. This represents the strongest evidence to date implicating DLX5 in the pathogenesis of SHFM and suggests a dual role for DLX5 in limb development.
Acknowledgments
We thank the family for their enthusiastic participation. We thank the Sequencing and Genotyping Core Facilities at KFSHRC for their help. This work was supported in part by a KACST grant 09-MED941-20 and Dubai-Harvard Foundation for Medical Research Collaborative Grant to FSA.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
Funding This study was supported by KACST, grant number 09-MED941-20 and Dubai-Harvard Foundation for Medical Research Collaborative Grant.
Competing interests None.
Patient consent Obtained.
Ethics approval Ethics approval was provided by IRB at King Faisal Specialist Hospital and Research Center.
Provenance and peer review Not commissioned; externally peer reviewed.