Article Text

Abstract
Background The lethal group of short-rib polydactyly (SRP) includes type I (Saldino-Noonan; MIM 263530), type II (Majewski; MIM 263520), type III (Verma-Naumoff; MIM 263510) and type IV (Beemer-Langer; MIM 269860). Jeune and Ellis-van Creveld dysplasias also used to be classified in the SRP group. Recently, mutations in a gene encoding a protein involved in intraflagellar transport, IFT80, have been identified in 3/39 patients with Jeune dysplasia but no extraskeletal manifestation.
Methods Because of clinical and radiological similarities between Jeune dysplasia and the other lethal types of SRP, the authors decided to investigate IFT80 in a cohort of fetuses with the lethal forms of SRP (Majewski, Verma-Naumoff and Beemer-Langer) and antenatally diagnosed cases of Jeune dysplasia. Fifteen fetuses were identified. A double-molecular approach was adopted. For consanguineous families and for those with recurrent sibs, a haplotype analysis around the gene locus was first performed, and, for the others, all the coding exons of IFT80 were directly sequenced.
Results Using the haplotype approach for two families, the authors excluded the IFT80 region as a candidate for them. Direct sequencing of IFT80 in the other 13 cases showed a G-to-C transversion in exon 8 (G241R) in only one SRP case closely related to the type III phenotype.
Conclusions The findings show that mutations in IFT80 can also be responsible for a lethal form of SRP and provide the molecular basis for the Jeune-Verma-Naumoff dysplasia spectrum.
- Short rib-polydactyly dysplasia
- IFT80
- ciliopathy
- Verma-Naumoff dysplasia
- Jeune dysplasia
- diagnosis
- genetics
- clinical genetics
- molecular genetics
Statistics from Altmetric.com
- Short rib-polydactyly dysplasia
- IFT80
- ciliopathy
- Verma-Naumoff dysplasia
- Jeune dysplasia
- diagnosis
- genetics
- clinical genetics
- molecular genetics
Introduction
Short-rib polydactyly (SRP) is part of a group of skeletal dysplasias that includes three less severe dysplasias—Jeune (MIM 208500), Ellis-van Creveld (MIM 225500) and Barnes (MIM 187760)—and four lethal types—Saldino-Noonan/Verma-Naumoff (types 1/3), Majewski (type 2), Beemer (type 3) and Mohr-Majewski (type 4).1 The proposed international classification is not a consensus, however, and, more commonly, the lethal group of SRP includes the following types: type I (Saldino-Noonan; MIM 263530), type II (Majewski; MIM 263520), type III (Verma-Naumoff; MIM 263510) and type IV (Beemer-Langer; MIM 269860).2 3 In addition, several phenotypes with overlapping features have remained unclassifiable.3–5
The molecular basis of these dysplasias started to be understood early this century with the discovery of mutations in the EVC1 and EVC2 genes.6 7 More recently, Beales et al8 found mutations in IFT80 in three of 39 Jeune families studied. IFT80 is a gene that encodes a well-conserved protein involved in intraflagellar transport, and is essential for the development and maintenance of motile and sensory cilia.9 Clinical and radiological similarities between Jeune syndrome and the other lethal types of SRP prompted us to investigate IFT80 in fetuses with the lethal forms of SRP.
Here we describe the analysis of this gene in a cohort of 15 probands presenting lethal types of SRP.
Patients and methods
Patients
Fetuses with lethal SRP were identified in three different institutions: the Perinatal Genetic Program (UNICAMP, Campinas, Brazil), the Bone Dysplasia Clinic at the Genetic Department of the Necker Hospital, and the Unit of Fetopathology of the Robert Debré Hospital (Paris, France). The inclusion criteria consisted of clinical and radiological data for confirmation of the diagnosis and DNA samples from the proband. All the families involved provided written informed consent for molecular studies that were previously approved by each local research ethics committee.
Methods
All babygrams were re-examined by some of the authors (DPC, MLM, VCD) in order to confirm the diagnosis. For molecular study, we adopted a double approach. For the consanguineous families and/or for those with recurrent sibs, we began the investigation by haplotype analysis at the IFT80 locus. For the other families, we directly sequenced the coding exons of IFT80 in the affected fetuses. For both procedures we used genomic DNA extracted by standard procedures. For the haplotype study, we used four markers at the IFT80 locus on chromosome 3 (3q26.1): GT repeats, 161430708–161430737; TG repeats, 161436455–161436497; TG repeats, 161625505–161625535; TA, repeats 161690223–161690271, sequence available on request (http://genome.ucsc.edu).
The complete sequencing of the 19 coding exons including the flanking intronic sequences of the IFT80 was performed using 19 intronic primers (sequence available on request). The PCR products were purified and sequenced by the fluorescent dideoxy-terminator method on an ABI 3130 (Applied Biosystems, Foster City, California, USA) automatic sequencer. Data were analysed by the Sequencing Analysis software (Applied Biosystems).
Results
We selected 15 fetuses that fulfilled the inclusion criteria. Clinical data, family history, origin and ethnic background of these cases are summarised in table 1. All fetuses, except case 10, were diagnosed early by prenatal ultrasound examination showing shortness of both limbs and ribs. All except cases 1, 2, 3, 4 and 10 were products of terminated pregnancy. Autopsy was performed on all except four fetuses: cases 1, 3, 4 and 9. Three families reported parental consanguinity (cases 9, 14 and 15), and it was also suspected in case 2 (see case report). Familial recurrence was observed in three families (cases 1, 11 and 15).
General clinical and familial data of the cases studied
Clinically, all patients presented disproportioned short stature with short limbs and narrowing thorax. Polydactyly was observed in nine cases (1, 2, 4, 7, 8, 9, 10, 14, 15). Other clinical features are summarised in table 2.
Main findings of external and internal examinations of the fetuses
Karyotypes, investigated in 10 cases (1, 2, 3, 4, 5, 6, 7, 8, 9, 11), were normal (table 3). Although typical radiological features were observed for each case, less severe skeletal findings were observed in one case of Jeune syndrome (6) and in three Verma-Naumoff cases (7, 8, 11). Typical radiological features observed for the different types were: (1) shortness of limbs and ribs in all cases; (2) smooth ends of the tubular bones, ovoid shape or absent tibia and pre/postaxial polydactyly of the four limbs in SRP type II; (3) metaphyseal irregularity of the long bones and trident acetabular roof in SRP type III; (4) narrowing of the thorax and typical trident acetabular roof in Jeune dysplasia; (5) smooth ends of the tubular bones and bowing of forearm bones in SRP type IV. We also observed some particularities of the hands and feet bones, ranging from severe hypoplasia to the absence of ossification, in the more severe cases of SRP type III (cases 2 and 3) and also in some cases of SRP type IV (cases 4, 10, 14).
Main radiological findings and karyotype of the cases studied
Of the identified families, the haplotype analysis of two (cases 9 and 11) allowed us to exclude the region of the IFT80 gene as a candidate.
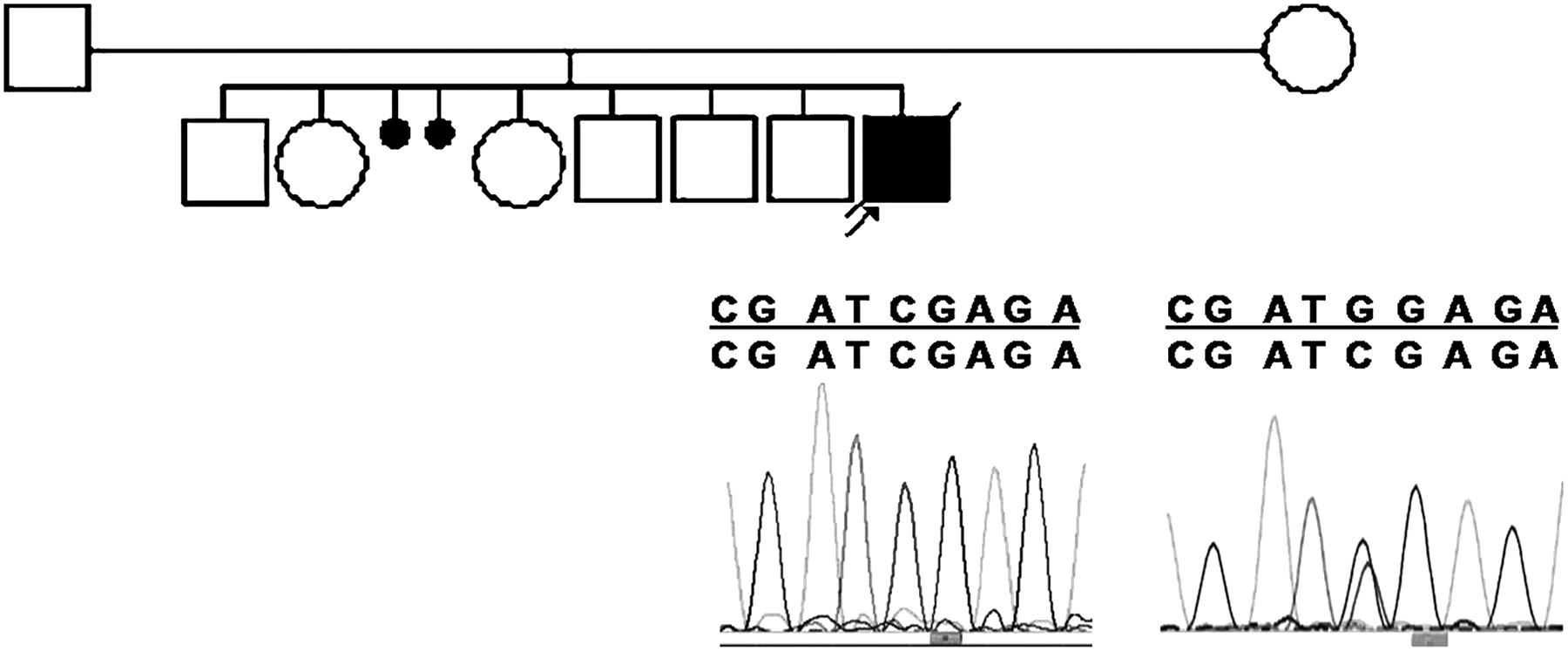
Sequencing of the 19 coding exons in the other 13 cases revealed a G-to-C transversion in exon 8 in case 2 (figure 1). This G241R mutation occurred in a well-conserved amino acid that is ‘predicted to be probably damaging’ in accordance with the alignment of almost 100% of species (http://genetics.bwh.harvard.edu/pph). Moreover, we did not find this mutation in 340 control chromosomes (180 European and 160 Brazilian), underlining that it should be a pathogenic mutation and not a common polymorphism in the human population.10 We also found a homozygous substitution (–40 C/T) in the non-coding region of exon 2 in the same patient. Sequencing of exons 2 and 8 of the mother showed that she was heterozygous for the two homozygous mutations found in her son (exon 2: –40 C/T; exon 8, base G1213C). Unfortunately, the father refused molecular analysis. The observed mutation in exon 2 appears to be a newly recognised polymorphism, because the heterozygous state was also found in one of the 58 controls examined (39 European and 19 Brazilian).
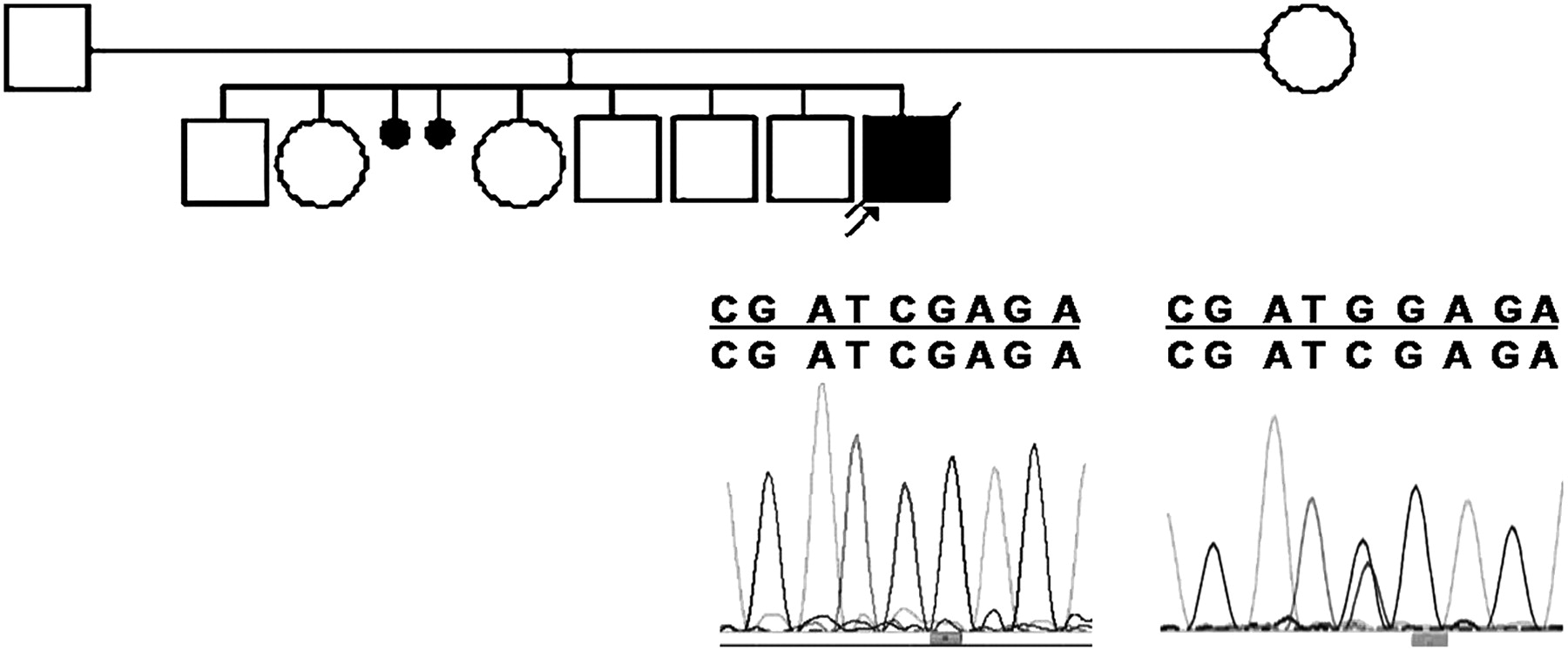
Genealogy of case 2 with partial electropherogram of exon 8 showing homozygous and heterozygous mutations G1213C (G241R) in the proband and his mother, respectively.
Report of case 2
The proband was a male stillborn, the 9th child of young parents with African black and Latin-European ancestries. Albeit denied, parental consanguinity was strongly suspected because both parents were from two close and very small cities of the Brazilian Northeast. This region is well known for its high inbreeding rate.11 The mother reported two miscarriages after the 2nd child (figure 1). Prenatal ultrasound examination at the third trimester showed short limbs, ascites, umbilical hernia and dorsolumbar scoliosis. Physical examination soon after delivery at 35 weeks showed a disproportioned fetus (height 36 cm; arm span 21 cm) with very short and curved limbs, slight trigonocephaly, submucous cleft palate, short and broad neck, narrow thorax, omphalocele, micropenis, small hands and feet with postaxial polydactyly on the hands and feet showing seven toes with duplication of the 1st and 2nd toes, and complete syndactyly between the duplicated digits. Minor facial dysmorphism included a high frontal, short and broad nose, very long and smooth philtrum, thin upper lip, isolated inferior central teeth and multiple oral frenulae. The babygram (figure 2A,B) revealed very short ribs with spreading ends, generalised platyspondyly, coronal cleft of lumbar vertebrae, abnormal scapulae, broad ischium, trident appearance of the pelvis, shortness of all long bones with metaphyseal irregularity and femur with bone spurs extending longitudinally from the medial and lateral segments of their metaphyses, giving a ‘banana-peel’ appearance. The long bone of the forearms and legs was very abnormal and short. Calcanei were duplicated (multiple), and the bones of the hands and feet were very hypoplastic, under-modelled and, sometimes, unossified (figure 2C,D). Autopsy findings showed: pulmonary hypoplasia, atrioventricular septal defect, enlarged thymus and intestinal malrotation. The placenta showed delayed maturation, with oedema and irregularity of the villi. Sections of the growth plate of the distal ends of the femur showed convexity and irregularity of the metaphyseal line, and a thick band of periosteal membranous ossification beyond the level of the physis. The growth plate was extremely disorganised and irregular, with shortness of both the proliferative and hypertrophic zones. However, the resting zone appeared normal. The bone trabeculae were thick and poorly organised with an excess of cartilage residua (figure 2E, F).

{kind=link}
{kind=link}
Babygram (A) with details (B–D) and section of the growth plate (E, F) of case 2. For description, see the text.
Discussion
Since the early 1970s, the phenotype characterised by disproportioned short stature with extremely short ribs and polydactyly has intrigued clinicians because of its clinical and radiological heterogeneity and the sometimes great difficulty or impossibility of making a correct clinical diagnosis.2–5
The discovery of the involvement of the IFT80 gene in Jeune dysplasia8 has placed this syndrome in the emerging class of human genetic disorders known as ciliopathies.9 Cilia and flagella are vital, specialised and evolutionarily conserved organelles that are broadly distributed in the tissues.9 12 These organelles consist of a basal body, which remains below the cellular surface, and an axoneme composed of internal microtubules extending through the cellular membrane where it develops chemosensors or mechanosensors functions.12 Mutations in the genes involved with the structure or function of the cilia are known to provoke a great number of different Mendelian disorders that share some common phenotypic features such as retina degeneration and polycystic kidney disease or, less commonly, anosmia, ataxia, liver fibrosis, cardiac defects, infertility, obesity, central nervous system abnormalities and skeletal dysplasia.8 9 12
IFT80 is one of the polypeptides of complex B of anterograde intraflagellar transport (IFT) mapping at 3q24-26.1. IFT is considered to be the process by which the so-called IFT proteins are transported together with other protein particles along the axoneme in both directions.12–17 Previously called WDR56, IFT80 has 20 exons and encodes a 777-residue protein containing seven WD40 domains.8
In the present series of 15 fetuses with SRP, we identified mutations in only one case. This mutation (G241R) in exon 8 is responsible for a change in a well-conserved amino acid region, suggesting that a partial loss of IFT80 function is the cause of the phenotype. The fetus in which we found the mutation presented a lethal form of SRP, with radiological findings compatible with SRP type III or Verma-Naumoff. Parental consanguinity in our case was strongly suspected because the parental families came from geographically close, small cities of the Brazilian Northeast. We have previously observed that part of the high parental consanguinity rate found in the Campinas region (∼3%), where the family lives, can be explained by migration from the Northeast region (unpublished data).
Beales et al8 highlighted three different mutations in four consanguineous patients with no extraskeletal manifestations—two missense in exons 4 and 19, and one in-frame deletion in exon 15—also suggesting a partial loss of IFT80 function.
In spite of some debate about classification of the lethal forms of SRP, type III or Verma-Naumoff SRP is characterised by a markedly narrow thorax, platyspondyly, coronal clefts, abnormal pelvis with a trident appearance and gross changes in the long bone, with metaphyseal irregularity and longitudinal spurs notably in the femur.18 19 These radiological features can range from moderate18 20 21 to severe skeletal involvement.18 22 23 Visceral malformations are often observed in this type of SRP; however, polydactyly is not a constant finding, but, when present, is usually postaxial.18 19 22 24 Although the radiological findings of the case in which we found the mutation are typical of a severe Verma-Naumoff SRP phenotype, preaxial polydactyly on the feet does not fit with clinical features of this type of SRP. A comparable case was reported by Young et al,23 in spite of the postaxial, rather than preaxial, polydactyly of hands and feet described in that fetus. As in other similar reported observations, these authors suggested a compound between Verma-Naumoff and oral-facial-digital syndrome (OFD) type II under the hypothesis of compound heterozygosity for two or more mutations.23 25 However, this suggestion is not consistent with the single mutation found in the fetus reported here. In fact, the ubiquitous character of the cilium12 makes it easier to assume a single mutation given a complex, pleiotropic and unexpected phenotype. Despite the fact that preaxial polydactyly has not been reported in Verma-Naumoff SRP, several authors include SRP types I and III as a single type and refer to preaxial polydactyly as an occasional finding.1 2 26 Moreover, preaxial polydactyly was also described by Majewski et al25 in a newborn (case I) presenting a phenotype compatible with Jeune dysplasia.
Even though apparently distinct from each other, Jeune dysplasia and Verma-Naumoff SRP exhibit radiological and histological similarities, suggesting that they could be variations of the same condition.23 25 Likewise, intrafamilial variation also supports the hypothesis that the two phenotypes are variants of the same pathological spectrum.20
The present results provide the genetic basis for the Jeune-Verma-Naumoff dysplasia spectrum and confirm the inclusion of these syndromes among the ciliopathies. Owing to overlapping of both clinical and radiological features among all SRP types, other cilium genes could also be involved in this group. Reinforcing this, two recently published reports showed mutations in a gene encoding a protein involved in retrograde transport in the cilium, associated with both Jeune dysplasia and SRP type III.27 28 In the first report, the authors identified DYNC2H1 mutations in three families diagnosed as having SRP type III,27 while in the second report mutations in DYNC2H1 were found in two fetuses with SRP type III and in three patients affected by Jeune dysplasia.28 Of note, in all presented cases of SRP type III with mutation in DYNC2H1, the radiological features are relatively moderate.27 28
On the basis of our results and these two recent published reports, we can speculate that, while DYNC2H1 mutations are associated with the less severe phenotype of SRP type III,27 28 mutations in IFT80 are very rare, until now observed in only four consanguineous families, and also responsible for the very severe SRP type III phenotype (present case).
In conclusion, the results of this investigation show that mutation in IFT80 can also be responsible for a lethal form of SRP and provide one of the molecular bases for the Jeune-Verma-Naumoff dysplasia spectrum. These findings also suggest that the other phenotypes of SRP may also result from cilium disruption.
Acknowledgments
We are indebted to the families who participated in the study. We also thank Dr Mauricio T Sakata for providing supplementary data from the Brazilian families.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Footnotes
Funding This work was supported by grants from Fapesp (98/16006-6), CAPES (0603/08-2) and PHRC (AOM06031).
Competing interests None.
Ethics approval This study was conducted with the approval of UNICAMP, Campinas, SP, Brazil, Hôpital Necker, Paris, France, Hôpital Robert Debre, Paris, France and Children's Hospital at Westmead, Sydney, Australia.
Provenance and peer review Not commissioned; externally peer reviewed.