Article Text

Abstract
Silver–Russell syndrome (SRS) is a heterogeneous disorder characterised by severe intrauterine and postnatal growth retardation, limb and body asymmetry, a typical facial appearance and less common dysmorphisms. Recently, epimutations and maternal duplications affecting the short arm of chromosome 11 have been shown to have a crucial role in the aetiology of the disease. Disturbances in the same genomic region cause the overgrowth disorder Beckwith–Wiedemann syndrome (BWS). In BWS, mutations in the telomeric as well as in the centromeric imprinting centres (ICR1 and ICR2) in 11p15 can be observed. In SRS, methylation defects in the imprinted region in 11p15 were considered to be restricted to the telomeric ICR1. They can be detected in about 30% of patients. This article reports on the first patient with SRS with a cryptic duplication restricted to the centromeric ICR2 domain in 11p15. The maternally inherited duplication in this patient included a region of 0.76–1 Mbp and affected the genes regulated by the ICR2, among them CDKN1C and LIT1. This study provides evidence for a role for this imprinting centre in the aetiology of SRS and shows that SRS presents a picture genetically opposite to that of BWS.
- BWS, Beckwith–Wiedemann syndrome
- ICR, imprinting centre region
- OFC, occipitofrontal circumference
- SRS, Silver–Russell syndrome
- STR, short tandem repeat
Statistics from Altmetric.com
- BWS, Beckwith–Wiedemann syndrome
- ICR, imprinting centre region
- OFC, occipitofrontal circumference
- SRS, Silver–Russell syndrome
- STR, short tandem repeat
Silver–Russell syndrome (SRS) is a mostly sporadic disorder characterised by severe intrauterine and postnatal growth retardation (<3rd percentile), clinodactyly V, asymmetry, typical craniofacial features including frontal bossing and a triangular face, and some other less common features. The disease is genetically heterogeneous: in about 10% of cases, maternal uniparental disomy of chromosome 7 can be identified.1 Chromosomal rearrangements have been described several times, among them disturbances affecting chromosome 11.2,3 In these cases, the complete imprinted region 11p15 was always involved. Chromosomal rearrangements, paternal uniparental disomy and epigenetic variations in this locus are well known to cause Beckwith–Wiedemann syndrome (BWS), a disease that is mainly characterised by overgrowth. Recently, similar but opposite epigenetic changes have been identified in patients with SRS: epigenetic variations in 11p15 account for about 30% and maternal duplications for about 4% of affected individuals.3–5 The imprinted region in 11p15 consists of two imprinting centre regions (ICRs): the telomeric ICR1 regulates the expression of IGF2 and H19, whereas the centromeric ICR2 controls the expression of CDKN1C, LIT1 (KCNQ1OT1) and several other factors.
In contrast with BWS, in which a preponderance of ICR2 epimutations can be observed, methylation defects in patients with SRS seem to be restricted to the telomeric ICR1.3–5 However, in mice, a targeted deletion of the paternal ICR2 copy results in growth retardation.6
Here, we report on the first patient with SRS with a mutation restricted to the ICR2 region.
CASE REPORT
Our patient was the first child of healthy unrelated parents. Owing to a deceleration of heart sounds, the child was delivered at 33 weeks’ gestation by caesarean section. Length at birth was 37 cm (−5.7 standard deviation (SD)), weight was 1240 g (−2.5SD), and occipitofrontal circumference (OFC) was 27.5 cm (−2.1 SD). The patient was fed intermittently via a gastrostomy tube because of prolonged feeding difficulties. At age 1 year, his height was 63.5 cm (−5.25SD), weight was 5350 g (−5.1SD) and OFC was 43 cm (−3.5SD). He was referred for genetic counselling at age 2.5 years with a length of 77 cm (−5.6SD), a weight of 7800 g (−3.3SD) and an OFC of 46.5 cm (−2.5SD) (fig 1). As dysmorphic signs, he presented a prominent forehead, a triangular face with a protruding philtrum, slightly downturned corners of the mouth and a small chin, posteriorly rotated large ears and clinodactyly of the fifth finger. He showed no evidence of lateral asymmetry. He was noted to have excessive sweating and a developmental delay: rolling over was achieved at 11 months, he crawled at 13 months and walked without support at the age of 2 years. His speech development was considerably retarded: at the age of 2.5 years, he spoke only single words. He is hyperactive and has attention deficit. His father’s height is 180 cm and his mother’s height 156 cm. Both were phenotypically normal. The maternal grandfather was 170 cm, the grandmother was 165 cm and the two sisters of the mother were 170 cm. Family history was unremarkable. Cytogenetic visible disturbances as well as maternal uniparental disomy 7 had been excluded before.

Patient at the age of 2.5 years with his mother. Parental/guardian informed consent was obtained for publication of this figure.
MATERIAL AND METHODS
DNA from the patient and his family was extracted from peripheral blood lymphocytes according to standard procedures. Duplication analysis was initially performed by short tandem repeat (STR) typing. Markers were localised within or close to the BWS region (table 1). After polymerase chain reaction, the alleles were visualised by an automated ABI377 sequencing system (ABI, Darmstadt, Germany). Information on primers and physical order of the markers were obtained from the Genome Database and the University of California at Santa Cruz browser (www.gdb.org;www.genome.ucsc.edu).
Results of short tandem repeat typing in the patient and his family
To further confirm the STR results, methylation analyses were carried out. In the case of the centromeric ICR2, bisulphite-treated DNA was amplified by polymerase chain reaction using primers localised in the differentially methylated region, followed by digestion with BstU1.7 Fragments were visualised by an automated ABI377 sequencing system, and methylation indices were calculated. Epimutations in the telomeric ICR1 were excluded by methylation-sensitive Southern blot analyses4: 10 µg of genomic DNA was digested overnight with HpaII and RsaI; the subsequent electrophoresis was run on a 1.2% agarose gel overnight. After capillary blotting, the nylon membranes were hybridised with a probe generated by polymerase chain reaction, and the digoxigenin-labelled fragment was detected according to standard protocols. The methylation index of each sample was calculated after densitometric measurement of autoradiographs using a GelDoc2000 system (BioRad, München, Germany).
For expression studies, RNA was isolated from immortalised lymphocytes from the patient and his mother. We then analysed a 12 bp (PAPA) deletion polymorphism in the CDKN1C gene.
Chromosome preparations were obtained from peripheral blood lymphocytes. Fluorescence in situ hybridisation was carried out using a whole chromosome painting probe for chromosome 11 (Quantum/Appligene, Illkirch, France) and a BAC clone mapping to the region of interest (RP11–66E9) (table 1).
RESULTS
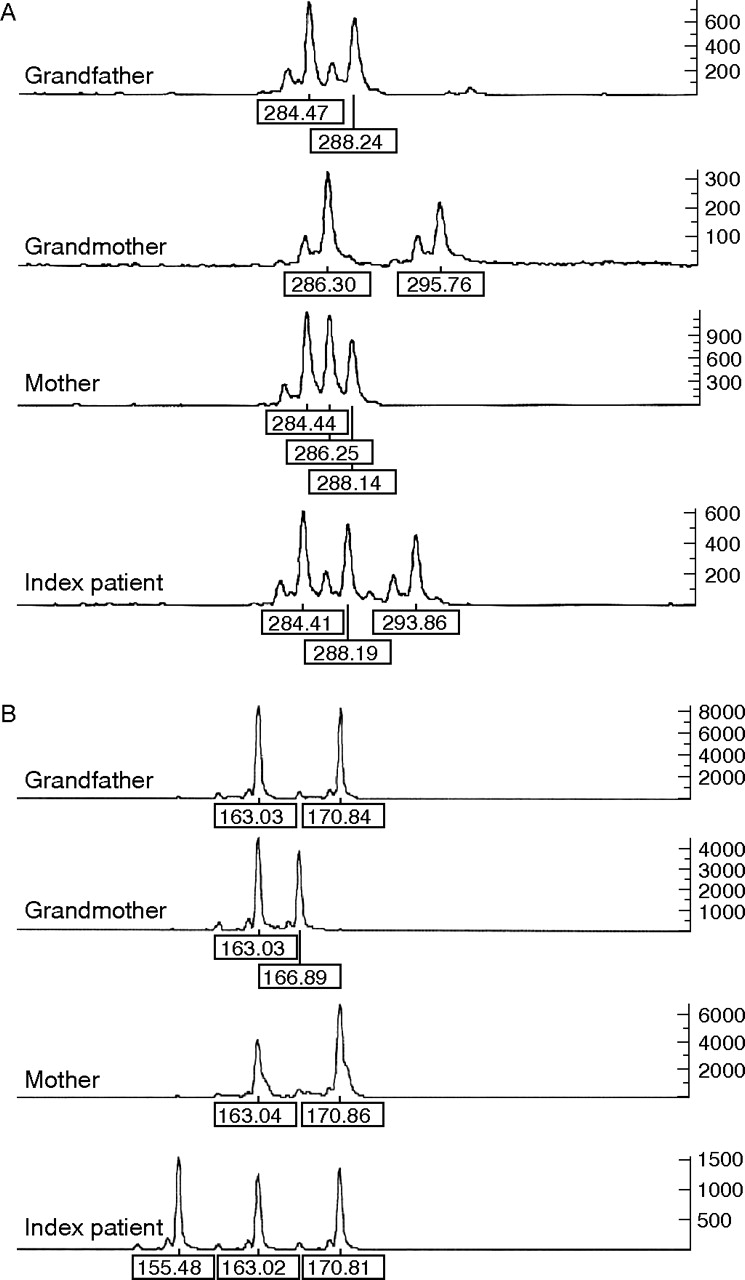
Typing of STR markers in our patient with SRS and his family yielded a mother-to-son transmission of a 0.76–1.0 Mbp duplication in 11p15 (table 1).
Although analyses of the grandparental DNA samples resulted in normal STR patterns, typing of the mother of our patient showed duplication of the paternal 11p15 material. Maintenance and reduction in paternal heterozygosity was observed (table 1). Interestingly, the allelic patterns in the index patient were consistent with a grandpaternal as well as with a grandmaternal contribution to the rearrangement (figs 2 and 3).

Example of short tandem repeat typing for marker (A) 11–2357 and (B) 11–3117. For both markers, three alleles could be detected in the patient, whereas typing of the mother showed maintenance of grandpaternal heterozygosity in the case of marker 11–2357 but reduction in homozygosity for marker 11–3117.

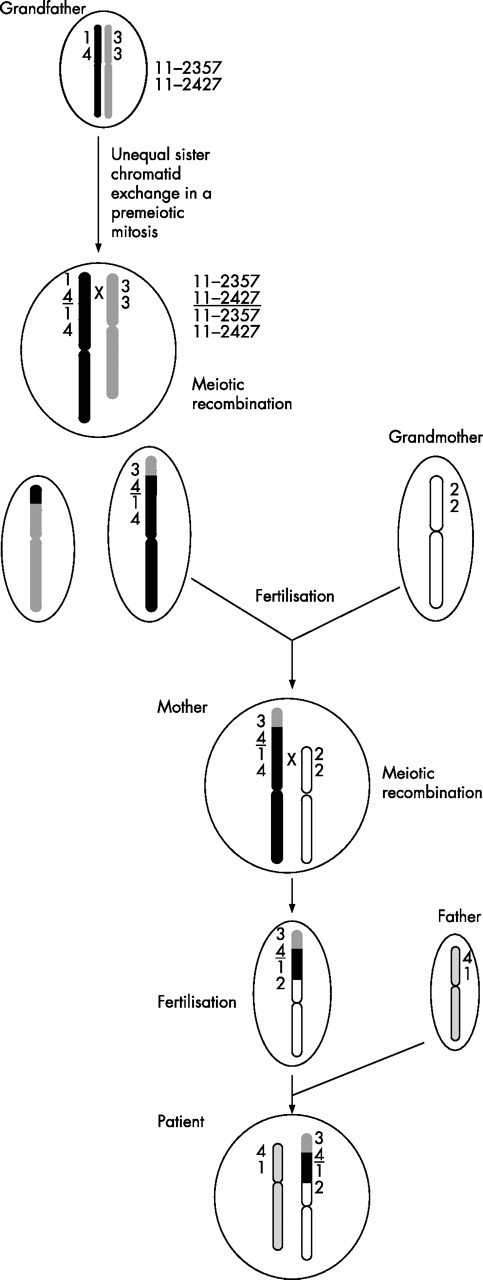
Schematic presentation of the assumed formation mechanism of the imprinting centre region (ICR2) duplication in 11p15 in our patient and his family. Further explanations are given in the text. Only the short tandem repeat typing results of the “key” markers 11–2357 and 11–2427 are shown.
By fluorescence in situ hybridisation analysis with a BAC clone mapping to 11p15, a stronger intensity of the fluorescence signal was observed on one chromosome 11 in each analysed mitosis in the patient and his mother. In interphase cells, elongation of the signal in one chromosome was detectable.
The duplicated region was restricted to the centromeric ICR2 and completely enclosed the maternally expressed genes KCNQ1, CDKN1C, TSSC5/SLC22A8 and TSSC3/PHDLA2 as well as the paternally expressed sequence LIT1 (KCNQ1OT1).
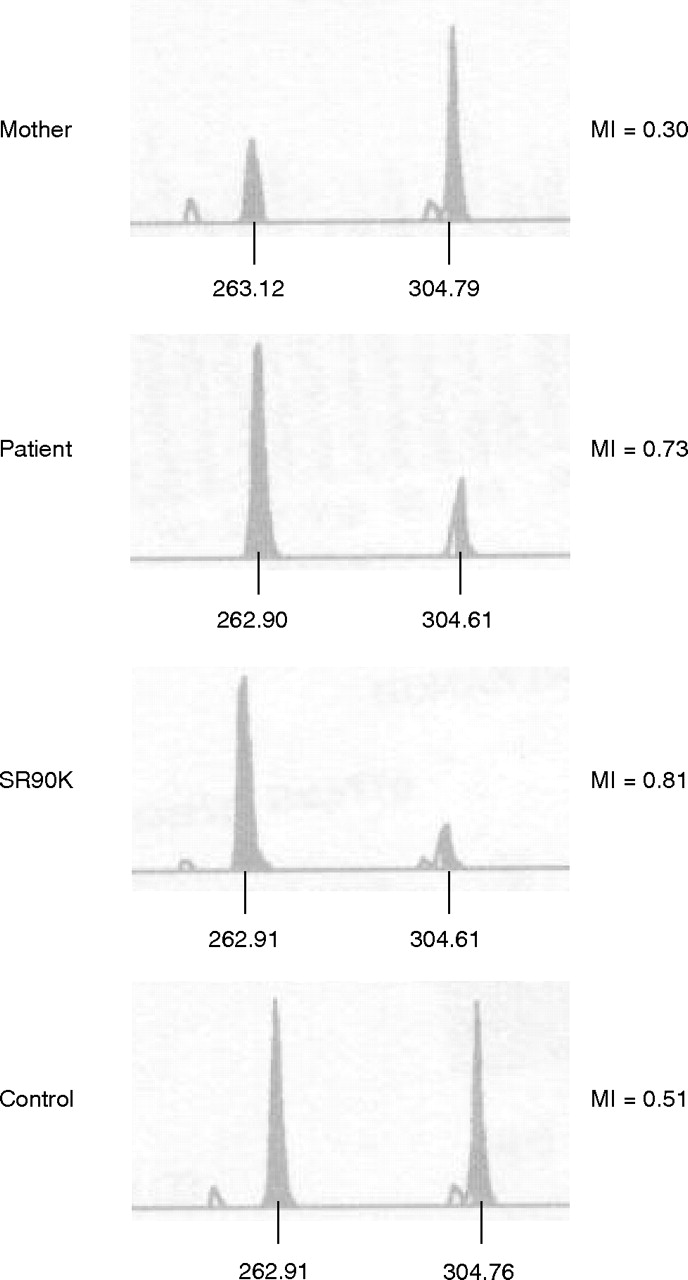
A methylation-specific restriction enzyme assay for ICR2 confirmed these findings (fig 4):7 we observed a strong increase in the mother’s unmethylated paternal allele, but in the patient the methylated maternal allele was stronger. In contrast, methylation analysis of ICR1 by Southern blotting showed normal results. We thus excluded an epigenetic methylation defect as well as a duplication in this region.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Methylation analysis of the imprinting centre region (ICR2) locus according to Cooper et al.7 The peak at about 263 bp represents the maternal methylated allele, whereas that at about 304 bp represents the paternal unmethylated allele. Methylation indices (MI) are given on the right side. SR90K, carrier of a maternal duplication 11p15.
At the genomic DNA level, the duplication was confirmed by analysis of the 12 bp (PAPA) deletion polymorphism in the CDKN1C gene. Different intensities of the two alleles in the patient and his mother were detectable (table 1). cDNA analysis showed maintenance of heterozygosity for the two alleles in the mother and the son, but without a difference in allele intensity. The finding in the patient’s cDNA is consistent with expression of the two different maternally inherited alleles. cDNA analysis of the mother showed expression of the maternal and also of the paternal alleles. However, the paternal origin of the duplication and the reported skewed inactivation of CDKN1C8 in human lymphocytes explain this observation.
DISCUSSION
With the identification of a cryptic duplication of the centromeric ICR2, we showed for the first time that mutations in 11p15 are not restricted to the ICR1 in patients with SRS and that the disease is indeed the genetically opposite clinical picture of BWS. The duplication enclosed the mainly maternally expressed genes KCNQ1, CDKN1C, TSSC5/SLC22A8 and TSSC3/PHDLA2, as well as the paternally expressed sequence LIT1 (KCNQ1OT1). CDKN1C is a negative regulator of cellular proliferation and growth. It has been hypothesised that the paternal CDKN1C allele is inhibited in many tissues by the untranslated RNA LIT1 which was also localised within the duplicated region in our patient. Although LIT1 is expressed paternally and its expression should be increased in the mother, this imbalance should not result in a generally altered phenotype, as LIT1 acts in cis and therefore only silences a supernumerary CDKN1C copy. In the index patient, the duplication is maternally inherited and should therefore result in overexpression of CDKN1C. On the basis of our cDNA data, we assume that he indeed has increased expression of CDKN1C, whereas the paternally derived duplication in the mother is not associated with a general stronger CDKN1C expression because of the regular inactivation of her paternal copies. However, in the mother’s lymphocytes, we detected partial imprinting of the duplicated segments, which might have an effect on the growth dependent of the affected tissue. Similar observations have been published by another group.8 This hypothesis is consistent with the mother’s relative small size. Finally, a contribution from altered expression of further genes in the duplicated region has to be considered.
The STR typing results indicate a complex formation mechanism in our patient and his family. On the basis of maintenance of and reduction in grandpaternal heterozygosity for single markers in the patient and a contribution of grandmaternal alleles in the duplication, we propose the following formation mechanism (fig 3).
The duplication itself should have occurred by unequal sister chromatid exchange in 11p15 in a premeiotic mitosis in the grandfather’s gonads. In the subsequent grandpaternal meiosis, a recombination between the rearranged chromosome 11 and its normal homologue between the markers 11–2357 and 11–2427 in the telomeric copy of the duplicated segment took place. As a result, STRs in the duplicated region in the daughter showed maintenance of (11–2357) and reduction in (11–2427) heterozygosity of paternal alleles. A further recombination occurred in the germline of the mother of our patient: here, a crossing over between the rearranged grandpaternal and grandmaternal chromosome 11p15 between markers 11–2357 and 11–2427 in the centromeric copy of the duplicated region can be observed; thus, our patient showed alleles inherited from his grandfather as well as from his grandmother.
In conclusion, we showed that the centromeric imprinting region ICR2 in 11p15 is involved in the aetiology of SRS, although epigenetic disturbances in ICR2 have so far not been reported. This case illustrates that SRS presents a picture genetically opposite to that of BWS, and that contrary genetic variations in 11p15 cause overgrowth as well as growth retardation.
Acknowledgments
We thank the proband and his family for cooperation and Dr B Schulze for the photographic documentation.
REFERENCES
Footnotes
-
Funding: This work was supported by the Doktor-Robert-Pfleger-Stiftung, Novonordisk Pharma GmbH, and a fellowship of the RWTH, Aachen, Germany (to NS).
-
Competing interests: None declared.
Further information: Parental/guardian informed consent was obtained for publication of figure 1.