Article Text

Abstract
Mutations in the human gap junction β-2 gene (GJB2) that encodes connexin-26 have been shown to cause non-syndromic sensorineural hearing loss (NSSNHL) at theDFNB1 locus on 13q11. Functional and genetic data regarding the disease causing potential of one particularGJB2 sequence variant, 101 T→C (M34T), have proven contradictory. In this study, we found the prevalence of the M34T allele in a cohort of white sib pairs and sporadic cases with NSSNHL from the United Kingdom and Ireland to be 3.179% of chromosomes screened. Significantly, we identified the first M34T/M34T genotype cosegregating in a single family with mid to high frequency NSSNHL. Screening a control population of 630 subjects we identified 25 M34T heterozygotes; however, no M34T homozygotes were detected. Surprisingly, the majority of M34T alleles (88%) were incis with a 10 bp deletion in the 5′ non-coding sequence. This non-coding deletion was also homozygous in the homozygous M34T subjects. Microsatellite analysis of flanking loci in M34T heterozygotes and controls does not define an extensive ancestral haplotype but preliminary data suggest two common alleles in subjects with the M34T allele. In summary, we provide data that support M34T acting as a recessive GJB2 allele associated with mild-moderate prelingual hearing impairment.
- GJB2
- connexin-26
- M34T
- hearing loss
Statistics from Altmetric.com
The gap junction β-2 gene (GJB2, MIM 121011), encoding connexin-26 (Cx26), is a member of the highly conserved connexin multigene family which encode integral membrane proteins that represent the structural component of gap junction intercellular channels.1 In 1997, mutations in GJB2 were shown to be responsible for non-syndromic autosomal recessive hearing loss at theDFNB1 locus.2 ,3 Subsequent analysis of GJB2 has led to identification of over 50 coding mutations (The Connexin-Deafness Homepage:http://www.iro.es/CX26deaf.html). One particular mutation, 35delG, accounts for up to 50% of autosomal recessive non-syndromic sensorineural hearing loss (NSSNHL) in some white populations.4 The frequency of the 35delG mutant allele varies in different populations, but may be as prevalent as 4% in particular ethnic groups.5 Two other commonCx26 mutations are 167delT and 235delC in cohorts of Ashkenazi Jews (4%) and Japanese (1%), respectively.6 ,7
The causal association of Cx26sequence variants detected in subjects with NSSNHL and the disease phenotype is based upon a number of standard criteria that include the segregation of the variant in affected subjects, its absence in controls, predicted effect on protein function, and data derived from functional analyses. However, the significance of one particular sequence variant of Cx26, 101 T→C (M34T), has proven difficult to assess given apparent contradictory genetic and functional observations.
The M34T amino acid substitution was first proposed as a dominant mutation (a candidate for the DFNA3 locus) from the analysis of a single family in which palmoplantar keratoderma and various forms of deafness were segregating.2 In this small family, two sisters with profound sensorineural hearing loss were heterozygous for the M34T allele. Subsequent analysis of family members showed the association of M34T with profound hearing loss only when M34T was in trans with a dominantGJB2 mutation, D66H.8Functional data generated using the Xenopus laevis paired oocyte assay showed that M34T Cx26 protein was a dominant disrupter of normal channel formation.9 Further support for its deleterious nature was shown using the mammalian HeLa cell expression assay. This showed that M34T Cx26 protein was targeted to the plasma membrane, but there was a significant reduction in oligomerisation and intercellular coupling owing to a net decrease in the amount of M34T containing connexons in the plasma membrane.10 Contradictory to these functional observations, the reports of the M34T allele in normal hearing subjects suggests the sequence variant does not function as a dominant, pathogenic variant in vivo (table 1). Thus, the M34T allele does not segregate with dominant hearing loss. Instead, the association of M34T in trans with alleles V95M, R184W, and 35delG in subjects with NSSNHL suggests a recessive mode of action.11 ,12
A summary of GJB2 studies assessing the prevalence of the M34T allele
Recently, a family consistent with recessive inheritance for NSSNHL has been described in which the M34T allele is segregating with a commonGJB2 recessive allele, 167delT.13 In this family, one subject was a 167delT/M34T compound heterozygote. The audiometric pattern in this person was said to be compatible with high frequency hearing loss.
In this study, we have screened affected sib pairs and sporadic cases from the UK and Irish population with moderate to profound prelingual NSSNHL for the M34T allele. In addition, a group of 630 controls (hearing status unknown) from the UK was also analysed. These analyses were performed in order to ascertain the phenotype of M34T/M34T homozygotes and also to assess the frequency of this variant in the British/Irish population.
Materials and methods
CRITERIA FOR INCLUSION IN THE STUDY
A cohort of 107 sib pairs and 66 sporadic cases with NSSNHL was collected from the British and Irish population. In all cases, clinical examinations were normal, thus excluding syndromal deafness, as were routine histories for acquired hearing loss caused by infection or ototoxic drugs. Ten ml of venous blood was collected from each patient and DNA extracted from peripheral blood lymphocytes using a standard non-organic extraction procedure. DNA was also analysed from 630 anonymised subjects resident in the UK (hearing status unknown) which formed our control population.
POLYMERASE CHAIN REACTION (PCR) AND SEQUENCING
PCR was performed in a 30 μl volume comprising 40 ng genomic DNA, 10 pmol of sense and antisense primer, 10 mmol/l Tris-HCl (pH 8.3), 50 mmol/l KCl, 1.5 mmol/l MgCl2, 0.1% (v/v) TritonX-100, a 200 μmol/l concentration of each dNTP, and 1 unit ofTaq DNA polymerase (Gibco BRL). Amplification conditions were as follows: 94°C for five minutes, 33 cycles of 94°C for 30 seconds, annealing temperature (table 2) for 30 seconds, and 72°C for 30 seconds, followed by 72°C for five minutes. PCR products were separated on a 1% (w/v) agarose gel and bands of expected size were excised and purified using the QIAquick Gel Extraction Kit (Qiagen). For sequencing, approximately 300 ng PCR product was used as template with the BigDye Terminator Cycle Sequencing Reaction Kit (PE Applied Biosystems). The products were visualised on an ABI377 DNA sequencer (PE Applied Biosystems).
Oligonucleotide primers used in GJB2 analysis. Primer pair M34T-F/R was used to screen for the M34T coding variant. Primer pairs Cx26-9F/R, Cx26-10F/R, Cx14-F/R, Cx26-16F/R, and Cx26-18F/R were used to amplify non-coding sequences and non-coding exon 1 (Genbank AF144321)
M34T IDENTIFICATION AND CODING SEQUENCE ANALYSIS
Primers M34T F/R were designed to amplify a 209 bp fragment ofGJB2 (table 2). PCR was performed in a 15 μl reaction volume with the following conditions: 94°C for two minutes, 30 cycles of 63°C for one minute, followed by 72°C for two minutes. An 8 μl aliquot of PCR product was used in a 15 μl restriction digest reaction with Hsp9211 according to instruction (Promega). The digest products were resolved on a 3% Metaphor gel (FMC Bioproducts) and compared to wild type (189 bp only) and heterozygous M34T (189 bp and 209 bp) controls. Confirmation of M34T genotypes and identification of compound mutations was performed by sequencing the entire Cx26coding region as described previously.2
CLONING OF GJB2 PCR FRAGMENTS
PCR products were cloned using the pGEM®-T Easy Vector System (Promega). PCR products were ligated overnight at 14°C and transformed into competent INValphaF' E coli(Invitrogen). Transformed cells were plated onto Luria-Bertani (LB)/ampicillin plates and incubated overnight at 37°C. Ten colonies were picked into 100 μl LB and incubated for one hour at 37°C. The LB broth was centrifuged, the pellet resuspended in 20 μl of TE (pH 8.0), and heated at 75°C for five minutes. A 2 μl aliquot was used in PCR reactions as described previously and the PCR product sequenced.
SCREENING NON-CODING SEQUENCES BY SSCP
The non-coding sequence of GJB2 was amplified using the primer pairs listed in table 2. Two μl of PCR product was restriction enzyme digested in a total volume of 20 μl according to the manufacturer's instruction (table 2, Promega and New England Biolabs) and 3 μl of the digest reaction was mixed with 3 μl of 95% (v/v) formamide, 20 mmol/l EDTA, 0.05% (w/v) bromophenol blue, and 0.05% (w/v) xylene cyanol. The sample was heated at 95°C for five minutes, cooled on ice for five minutes, and loaded onto a 12.5 % (w/v) non-denaturing polyacrylamide gel (GenGel Excel 12.5, Pharmacia Biotech). Electrophoresis was performed at 25 mA/14°C for 90 minutes (Genephor Unit, Pharmacia Biotech) and the DNA visualised following silver staining. Samples with a different electrophoretic mobility to known wild type controls were sequenced.
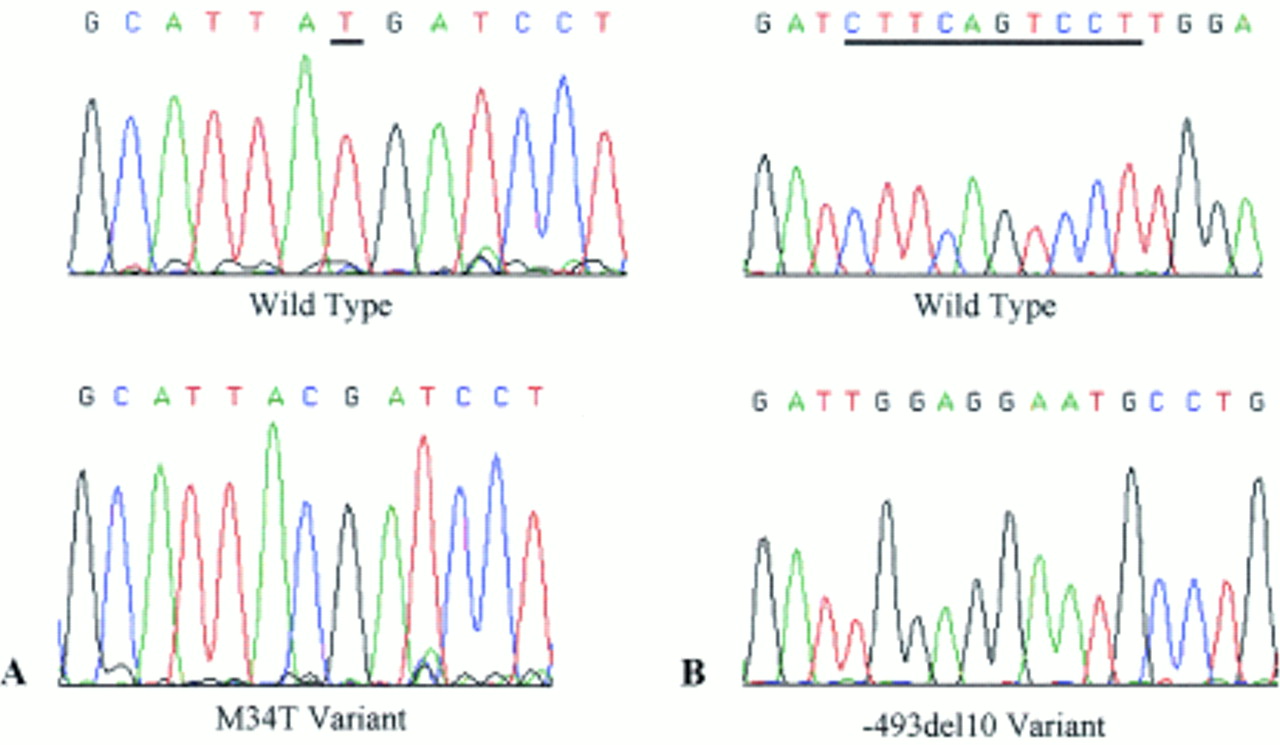
IDENTIFICATION OF THE −493DEL10 VARIANT
For each subject, the Cx26-14 fragment (383 bp) was PCR amplified and an 8 μl aliquot transferred to a 20 μl digest reaction with BglII restriction enzyme. Then 10 μl of digest product was visualised on a 1% agarose gel following electrophoresis for 30 minutes. In the absence of −493del10, the PCR product is cut into two fragments of 143 bp and 240 bp.
HAPLOTYPE ANALYSIS
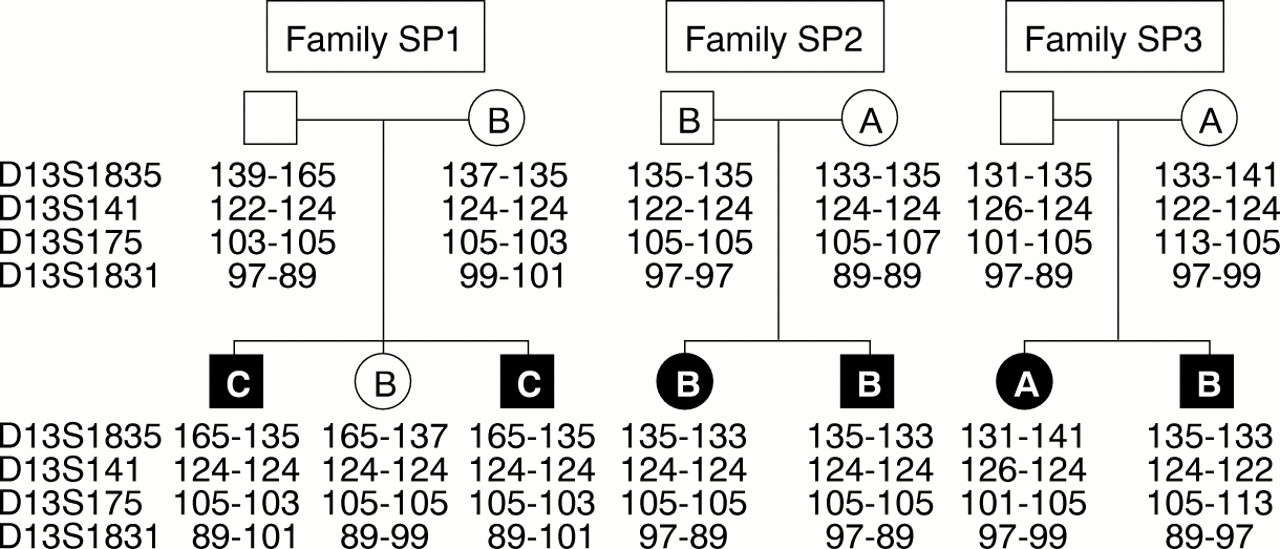
Genotyping markers D13S1835, D13S141, D13S175, and D13S1831 from 13q11 were used to generate haplotypes for subjects with the M34T allele and some family members. PCR product was visualised using an ABI 377 and analysed with Genotyper software according to instruction (PE Applied Biosystems).
CELL CULTURE AND RT-PCR ANALYSIS OF M34T/WT CELL LINES
Towards investigating the expression of the M34T allele, we screened DNA extracted from a number of anonymised primary keratinocyte cultures and cell lines for the M34T allele. M34T heterozygotes were identified in one primary keratinocyte culture and in four cell lines. These samples were also heterozygous for –493del10. Primary keratinocytes were prepared by enzymatic dispersion and cultured.14 Cell pellets for the cell lines A431 derived from epidermoid carcinoma, 833K derived from testicular tumour line, ASPC-1 derived from pancreatic adenocarcinoma, and XPG415 derived from skin fibroblasts (xeroderma pigmentosum group G) were obtained from Research Cell Services at the Imperial Cancer Research Fund. Total RNA was extracted using Trizol reagent (Life Technologies, UK). RNA (1 μg) was reverse transcribed to first strand cDNA by moloney murine leukemia virus reverse transcriptase and random hexamer (Perkin Elmer, UK) in 20 μl of reaction mixture, and 5 μl of RT product was applied to PCR using Taq DNA polymerase (Perkin Elmer, UK) with forward primer CAGCGCAGAGACCCCAACGC and reverse primer GACACGAAGATCAGCTGCAG (5′-3′). The PCR programme was 94°C for two minutes, followed by 35 cycles as follows: 94°C for 30 seconds, 60°C for 30 seconds, 72°C for 45 seconds, and completed for a final extension of 72°C for 10 minutes. RT-PCR products were checked on 1% agarose gel and sequenced using BigDye Sequencing chemistry (Perkin-Elmer, UK).
Results
The M34T allele was detected in seven of 107 sib pairs and three of 66 sporadic cases with NSSNHL. In three of the seven sib pairs (SP3, SP4, and SP5) the M34T allele did not segregate in both affected subjects and in two cases (SP6 and SP7) only one sib was available for testing (table 3). Significantly, we identified the first subjects homozygous (designated sib pair SP1) for the M34T sequence variant (table 3, fig 1A). The affected offspring presented with a mild-moderate prelingual bilateral hearing loss (fig 2). Their normal hearing sister was heterozygous for M34T. In addition, we screened 630 anonymous subjects (whose hearing status was not available) and did not identify any further M34T homozygotes. However, the M34T allele was detected in 25 of the 1260 (1.984%) chromosomes screened compared to a frequency of 3.179% in the NSSNHL cohort (table 1).
A summary of the sib pair (SP) and sporadic (S) cases with NSSNHL identified with M34T or −493del10 sequence variants. A or B after the SP prefix denotes sib 1 or 2; in some cases only one sib was available for screening

Sequence electrophoretograms taken from an ABI 377 representing (A) wild type and homozygous M34T traces and (B) wild type and homozygous −493del10 traces. The underlined nucleotide(s) in the wild type sequence denote the base(s) modified in the variant.

Tonal audiometric curves from both ears in one M34T homozygote identified in the study.
Sequence analysis of the coding region in M34T sib pair and sporadic cases indicated that two sib pairs (SP5A and SP6A) were compound heterozygotes with the 35delG mutant GJB2allele (table 3). The audiogram of subject SP6A showed a U shaped moderate NSSNHL. Subject SP5A has William's syndrome and is thought to have a mild-moderate sensorineural hearing loss. Subcloning the PCR fragments in these subjects established that both sequence variants were in trans. DNA from the second affected sib was available for analysis in one of the families (SP5B) and was shown to carry only the 35delG mutation. This subject had profound NSSNHL. Thus, M34T did not segregate with NSSNHL in this family. No other GJB2 mutations were detected in the NSSNHL M34T heterozygotes.
To extend the analysis of the genetic background of M34T gene heterozygotes with NSSNHL in which no secondGJB2 coding mutation was detected, we performed SSCP analysis of the non-coding exon 1 and 1.5 kb of sequence immediately 5′ to exon 1. In the seven M34T heterozygotes analysed, a heterozygous 10 bp deletion was identified in the non-coding upstream sequence (fragment Cx26-14), spanning nucleotides −493 to −502, relative to the transcription start site of exon 1 (−493del10). The −493del10 variant was subsequently detected on the basis that it abolishes aBglII restriction enzyme site. Extending this analysis to the remaining subjects with M34T (SP1, SP5, and SP6A) identified that two of the three also carried −493del10. The −493del10 variant was in a homozygous state in both M34T homozygous offspring (fig 1B). To assess the prevalence of −493del10 in other subjects with NSSNHL, we screened the remaining 100 sib pairs and 63 sporadic cases. In three of 100 (3.0%) sib pairs and two of 63 (3.2%) sporadic cases, the −493del10 variant was identified. Three of the four analysed M34T heterozygotes with mild-moderate NSSNHL from Australia were also heterozygous for −493del10. Two of them were compound heterozygotes with 35delG. In the control population, 22 of the 25 (88.0 %) M34T heterozygotes and 40 subjects (6.4%) without the M34T allele also carried −493del10. No additional −493del10 homozygotes were detected in the remaining subjects with NSSNHL or from the control population.
Our data suggest that there is linkage disequilibrium between M34T and −493del10. On this premise, we hypothesised that M34T is an ancestral mutation occurring on a GJB2 allele previously harbouring −493del10. To identify this proposed ancestral haplotype, four microsatellite markers spanning a <1 cM interval encompassing the GJB2 locus were used to genotype M34T heterozygotes and some family members (fig 3). In the homozygous M34T subjects, homozygosity was only observed with marker D13S141 (allele size 124 bp). Analysis of three other sib pair families identified a common M34T/−493del10 deletion haplotype at the loci D13S1835, D13S141, and D13S175 (allele sizes 135 bp, 124 bp, and 105 bp respectively). The frequency of this proposed haplotype was assessed in M34T/−493del10 deletion heterozygotes and then compared with a control group previously shown not to have M34T or −493del10 alleles. In 14 of 23 (60.9%) heterozygotes the proposed haplotype was possible; however, without other family members available for testing it is difficult to determine which alleles cosegregate with the M34T/−493del10 allele. Refining the haplotype to flanking markers D13S141 (124 bp) and D13S175 (105 bp) increases the number of M34T/−493del10 heterozygotes to 20 of 23 (87.0%). Analysis of the control group indicates that six of 32 (18.8%) subjects have the extended haplotype, increasing to 11 of 32 (34.4%) with the refined haplotype.
{kind=link}
{kind=link}
{kind=link}
Haplotype analysis of M34T/10 bp deletion families. Microsatellite markers D13S1385 and D13S141 are centromeric to GJB2 and D13S175 and D13S1381 are telomeric. D13S1385, D13S141, and D13S175 flank GJB2 in an interval of <500 kb, and D13S1381 maps more telomeric.24 ,25 Allele sizes are shown in bp. The letter assigned to a subject in the pedigree denotes the M34T/−493del10 GJB2 genotype: (A) wild type, (B) heterozygous M34T/−493del10, and (C) homozygous M34T/−493del10.
In addition, we investigated whether –493del10 had an effect on the transcription of Cx26, specifically that of M34T. RT-PCR was performed on cultured keratinocytes from a heterozygote for the M34T/−493del10 allele. In addition, four cell lines with the same genotype were also analysed. Direct sequencing of the resulting Cx26 PCR products showed that the M34T/−493del10 allele was expressed (data not shown).
Discussion
Investigation of the M34T mutant allele in sib pairs (6.5%) and sporadic cases (4.5%) with NSSNHL from the UK and Ireland has led to identification of the first M34T homozygote. Both sibs presented with mid to high frequency prelingual hearing loss supporting previous reports of M34T acting as a recessive GJB2hearing loss allele. Compound heterozygotes of M34T with 35delG were also identified; however, segregation analyses showed that M34T did not segregate with NSSNHL in one family. This observation could be explained by hearing loss being the result of a mutation in another hearing loss associated gene.
Genetic studies of GJB2 suggest that the M34T variant is more prevalent in the mid-west American (1 in 130 chromosomes), UK, and Irish (1 in 50.4 chromosomes) normal hearing populations (table 1), but has reduced frequency or absence in those of French, Spanish, Italian, and Japanese ethnic origin. These population data suggest that a single ancestral mutation event occurring originally in the UK or Ireland may account for these population differences in numbers of M34T heterozygotes. Indeed, the occurrence of the −493del10 variant in cis with M34T may represent a marker of this proposed founding allele. Microsatellite analysis using four markers, three of which map to within <500 kb ofGJB2, showed an association of M34T with D13S141 (allele size 124 bp) and D13S175 (allele size 105 bp). The markers D13S141 and D13S175 flank GJB2 and have been used previously to identify a common haplotype for the 167delT mutation.6 ,15 However, the homozygous M34T subjects identified in this study were only homozygous for the 124 bp allele at the D13S141 locus in addition to −493del10. The possibility of historical recombination events and/or microsatellite mutations masking a common haplotype may explain these observations. Extended analysis of M34T subjects with closer microsatellite markers or SNPs may show more conclusive evidence for a founding haplotype.
The role of −493del10 in GJB2 regulation is difficult to define. Recently, a single nucleotide substitution identified in the regulatory region of presenilin 1 was shown to reduce reporter gene expression by 30% in transiently transfected neuroblastoma cells.16 The GJB2regulatory region has been characterised to some extent in certain human cell lines and indicates that the 128 bp immediately upstream of exon 1 are important for gene expression.17 However, the −493del10 variant is positioned outside this putative regulatory region (−493 to −502). From analysis of cultured keratinocytes and cell lines, the M34T/−493del10 allele was expressed. This result suggests two possibilities for the mode of action of −493del10. Firstly, the deletion may only affect promoter activity in certain cell types such as the epithelial cells of the inner ear and not in the epidermis. However, the analysis used was not quantitative so subtle differences in expression would not have been identified. Alternatively, −493del10 is a neutral polymorphism that has little or no biological consequence on Cx26 mRNA and protein levels. In support of the latter is the identification of a few subjects heterozygous for M34T who did not have −493del10 but had normal hearing and a subject (SP6A) with moderate NSSNHL who was a compound heterozygote for M34T/35delG without –493del10.
A number of reports of 35delG (mild-moderate), 167delT (compatible with high frequency), V95M (degree of hearing impairment not known), R184W (mild), and now M34T (mid-high frequency), associated intrans with an M34T allele supports the recessive nature of M34T.11-13 Both 35delG and 167delT are frameshift mutations that truncate the Cx26 protein at codon 13 and 81 respectively. In vitro studies of Cx32have shown that truncation of the protein before amino acid 207 has deleterious effects on targeting to the membrane, oligomerisation, and channel function.18 Therefore, in 35delG/M34T and 167delT/M34T subjects, the frameshift mutant allele should have no or little effect in gap junction formation and function.18 On this premise, the M34T protein, which has been to shown to reach the membrane,10 is solely contributing to theCx26 associated gap junction channels in the cochlea. These data suggest that further research into the role of M34T and NSSNHL is required so that genetic counselling can be provided, particularly in the UK and Ireland which have a high M34T heterozygote frequency.
Acknowledgments
We would like to thank all the participants for taking part in the study. This work was supported by the NHS Research and Development Trust, Wellcome Trust, National Lotteries Charity Board through Defeating Deafness, the European Union Framework V, and a bequest from the Special Trustess of St Bartholomews' Hospital.