Article Text

Abstract
Neurofibromatosis type I (NF1) is an autosomal dominant disorder affecting 1 in 3000 people. The NF1gene is located on chromosome 17q11.2, spans 350 kb of genomic DNA, and contains 60 exons. A major phenotypic feature of the disease is the widespread occurrence of benign dermal and plexiform neurofibromas. Genetic and biochemical data support the hypothesis thatNF1 acts as a tumour suppressor gene. Molecular analysis of a number of NF1 specific tumours has shown the inactivation of both NF1 alleles during tumourigenesis, in accordance with Knudson's “two hit” hypothesis. We have studied 82 tumours from 45 NF1 patients. Two separate strategies were used in this study to search for the somatic changes involved in the formation of NF1 tumours. First, evidence of loss of heterozygosity (LOH) of the NF1 gene region was investigated, and, second, a screen for the presence of sequence alterations was conducted on a large panel of DNA derived from matched blood/tumour pairs. In this study, the largest of its kind to date, we found that 12% of the tumours (10/82) exhibited LOH; previous studies have detected LOH in 3-36% of the neurofibromas examined. In addition, an SSCP/HA mutation screen identified five novelNF1 germline and two somatic mutations. In a plexiform neurofibroma from an NF1 patient, mutations in bothNF1 alleles have been characterised.
- neurofibromatosis
- NF1gene
- somatic mutations
Statistics from Altmetric.com
Neurofibromatosis type 1 (NF1) is a common autosomal disorder affecting approximately 1 in 3000 people. TheNF1 gene, located at 17q11.2, has 60 exons spanning approximately 350 kb of genomic DNA.1-4 TheNF1 gene product, neurofibromin, is structurally and functionally related to the GTPase activating protein (GAP) family. This includes the yeast IRA1and IRA2 genes, which are known to downregulate p21rasactivity.5 The most highly conserved region of neurofibromin, the NF1-GAP related domain (NF1-GRD), is encoded by exons 20-27a of the NF1 gene.
A major phenotypic feature of NF1 is the widespread development of multiple neurofibromas.6 These are benign tumours derived from the connective tissue of nerve sheaths, particularly the endoneurium. Dermal neurofibromas represent discrete focal lesions of the nerve sheath, composed predominantly of Schwann cells and fibroblasts, but in addition contain axons, perineurial cells, mast cells, and extracellular matrix. Plexiform neurofibromas are usually more extensive as their growth occurs along the length of a nerve and may involve multiple fascicles. Histopathology indicates that plexiform neurofibromas usually contain multiple cell types typical of neurofibromas, but also contain a greatly expanded extracellular matrix.7 Although similar to dermal neurofibromas, plexiform neurofibromas do have the potential to become malignant.
Almost all reported NF1 mutations are predicted to lead to neurofibromatosis by the direct inactivation of the gene; this property coupled with its known role in downregulating the ras pathway has led to the hypothesis that NF1 is a tumour suppressor gene.8 Knudson's “two hit” hypothesis of tumourigenesis requires the biallelic inactivation of a specific gene for it to be considered as a tumour suppressor.9 Studies on a number of tumour suppressor genes have shown that the second (somatic) inactivating mutation often results from the loss of the chromosomal region containing the suppressor gene. The resultant hemizygosity for such a chromosomal region can be monitored by screening the patient's DNA with polymorphic markers from within the deleted region. Thus, a search for evidence of loss of heterozygosity (LOH) within a particular gene region should identify such deletions.
There have been few reports identifying the biallelic inactivation of the NF1 gene in a tumour. An initial study by Coleman et al 10 showed LOH in more than 36% (8/22) of the dermal neurofibromas they analysed from five unrelated NF1 patients, while Serra et al 11 found LOH in only 25% of the 60 neurofibromas they screened. In direct contrast, however, a recent study by Daschneret al 12 found little evidence for any LOH across the entire NF1 gene region. Only 2.6% of the 38 neurofibromas they examined showed a deletion.12
In this study, we have identified LOH in 12% of 82 neurofibromas screened from 45 patients using a panel of 13 markers. We also report the first case in which both the germline and somaticNF1 mutations have been identified in DNA from a plexiform neurofibroma. A somatic nonsense mutation was detected in exon 16 (R816X) on the normal NF1 allele and the constitutional sequence alteration was subsequently found to be a lesion in the obligate donor splice site GT dinucleotides (IVS4+1G→A) of intron 4.
Materials and methods
PATIENTS
Patients were diagnosed as being affected by NF1 according to the diagnostic criteria formulated by the National Institute of Health Consensus Conference (NIH CC) 1987.
TUMOURS
Our tumour panel includes DNA isolated from 82 NF1 specific tumours (74 dermal neurofibromas, three plexiform neurofibromas, two pilocytic astrocytomas, and three schwannomas) from 45 NF1 patients. The corresponding lymphocyte DNA was also available from these patients. Plexiform neurofibromas were sent from clinics with specialist interest in neurofibromatosis.
DNA EXTRACTION
Tumour samples were transported in tissue culture medium and contaminating tissues, including skin, fat, and hair, was carefully dissected away under the supervision of a pathologist.
DNA was extracted by incubating tumour tissue for 1.5 hours at 65°C in 10% SDS, tissue extraction buffer (5 mmol/l NaCl, 1 mol/l Tris, 0.5 mol/l EDTA, 10% SDS) and proteinase K. Phenol/chloroform extraction was carried out and the DNA precipitated by ethanol and 3 mol/l sodium acetate. Constitutional DNA was extracted from peripheral blood cells by conventional methods.13
PCR
Amplification of the DNA was carried out in a 12.5 μl volume containing 10 ng DNA, 12 pmol/l of each primer, 1.5 mmol/l MgCl2, 1.25 mmol/l each dNTP, 1 UTaq polymerase, and buffer (Qiagen). Reactions were heated to 94°C for five minutes and then subjected to 35 cycles at 94°C for 30 seconds, 50-58°C for 30 seconds, 72°C for 30 seconds, with a final extension at 72°C for 10 minutes. The NF1 primers used were as previously published.14 15
LOH STUDIES
A panel of 13 markers16-27 (eight intragenic and five extragenic to NF1) were used to screen the panel of 45 tumour and blood pairs (table 1) to examine LOH in tumour tissue.28
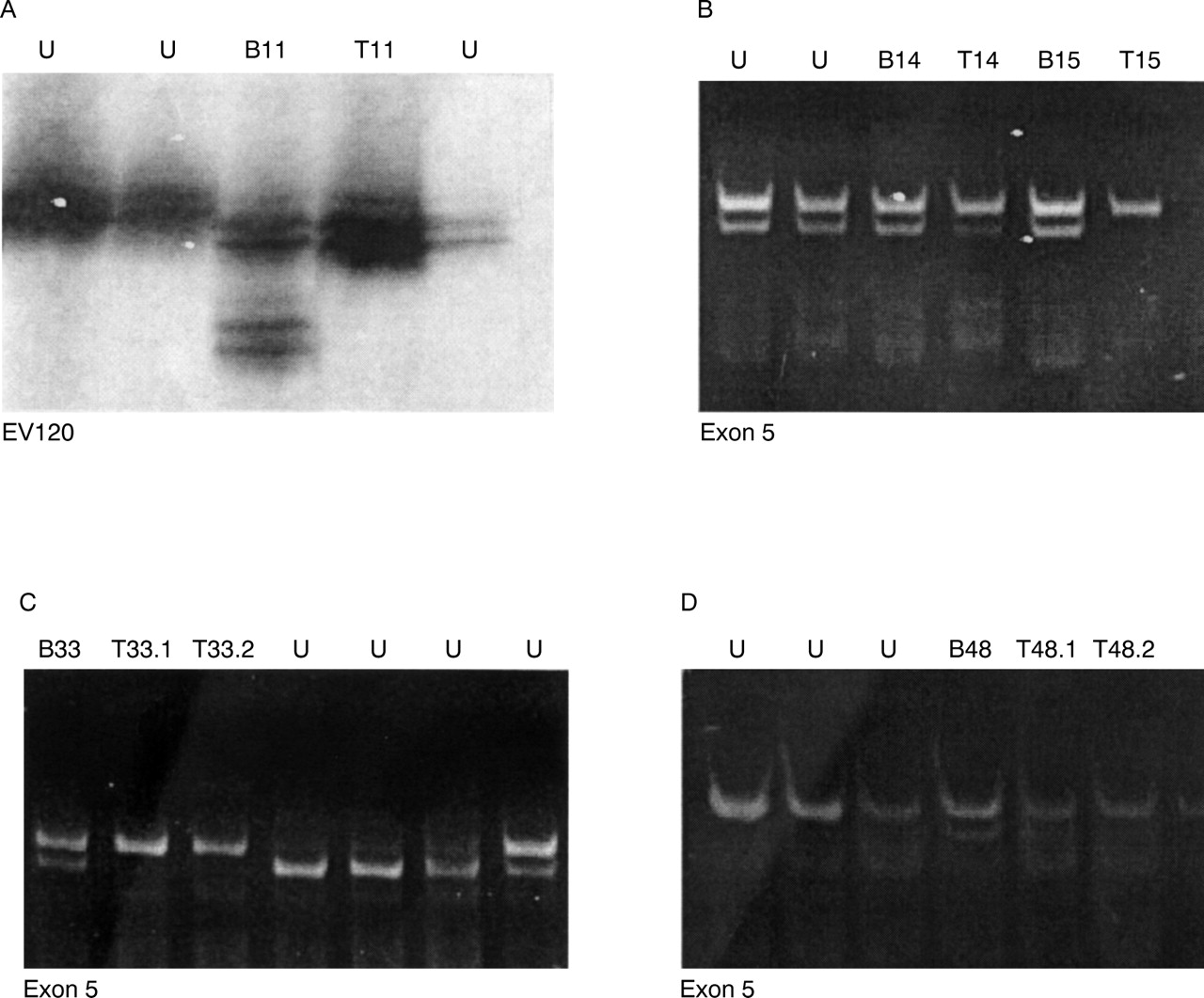
Results of LOH studies at NF1 and flanking polymorphisms in four patients, screened with eight intragenic and five extragenic markers. Black circles denote undeleted regions, white circles denote LOH, black squares denote uninformative loci, and ND denotes samples that would not PCR for that particular marker
HETERODUPLEX ANALYSIS (HA)
A total of 15-20 ng of amplified DNA (two PCR products were pooled to increase sample throughput) was mixed with 1 μl 0.1 mol/l EDTA, heated to 94°C for three minutes, and slowly cooled to 40°C over 40 minutes to allow heteroduplex formation. The reannealed product was loaded onto 24 cm × 33 cm × 0.8 mm gels and electrophoresed at approximately 5 W/hour until the marker dye front was at least 15 cm from the origin.29
SINGLE STRANDED CONFORMATION POLYMORPHISM (SSCP) ANALYSIS
Two μl of PCR product (8-10 ng DNA) was diluted 1:5 with loading buffer (95% formamide, 10 mmol/l NaOH, 0.25% bromophenol blue, 0.25% xylene cyanol) and heated to 94°C for three minutes. The product was loaded immediately onto 0.5 × MDE (Flowgen) gels and electrophoresed at room temperature for 50-150 Watt hours depending on the product size.
SILVER STAINING
Bands were visualised by silver staining. Gels were soaked in 0.1% silver nitrate for five minutes, rinsed briefly in distilled water, and then soaked in developing solution (1.5% sodium hydroxide, 0.01% sodium borohydride, and 0.15% formaldehyde) until bands appeared (five to 15 minutes). The bands were fixed by soaking in 0.75% sodium carbonate for two to five minutes and then rinsed briefly in water. The gel was transferred onto 3MM Whatmann paper and dried at room temperature. Aberrant bands were identified by their differing migration patterns.
SEQUENCING
Exons were amplified by cycle sequencing for 35 cycles using the Thermo Sequenase cycle sequencing kit from Amersham. Primers were labelled using [γP33]dATP (Amersham) and T4 polynucleotide kinase (Promega). Approximately 3 μl (12-20 ng) product was diluted 1:1 with loading buffer and run on polyacrylamide gels for 1.5-3 hours at 75 W/hour.
Results
Evidence for LOH of the NF1 region was investigated in the 45 tumour/blood pairs (table 1). A panel of eight intragenic and five extragenic DNA polymorphic markers was used.28 Ten out of 82 (12%) neurofibromas, isolated from seven separate patients, have shown evidence for LOH (fig 1). The end points of these deletions have not been defined and the constitutional NF1mutations have still to be identified.

LOH of the NF1 gene region: LOH seen in three patients. (A) Patient T11, LOH at marker EV120 RFLP in intron 27b. (B) Patient T15 and patient T14 both showing LOH at the exon 5 RFLP. (C) Two tumours from patient T33, and (D) two tumours from patient T48, both displaying LOH at the exon 5 RFLP. B=blood DNA, T=tumour DNA, U=other patient samples.
Exons 16-40 (29 exons) of the NF1 gene were targeted for investigation. This conserved region of the gene exhibits extended homology with the yeast IRA1 andIRA2 genes and is still the only domain of the protein to which any function has been ascribed and is involved in the downregulation of the p21 ras oncogene.7 The majority of exons (28/29) encompassing this region were screened in this study. Owing to problems associated with the variable quality and quantity of genomic DNA isolated from some tumours, approximately 1-5% of DNA samples proved to be refractory to mutational analysis. Five novel germline mutations and two somatic sequence alterations were found following the SSCP or HA mutational screen of DNA (table2).
Five germline and two somatic mutations identified following a screen of exons 16-40 using SSCP/HA
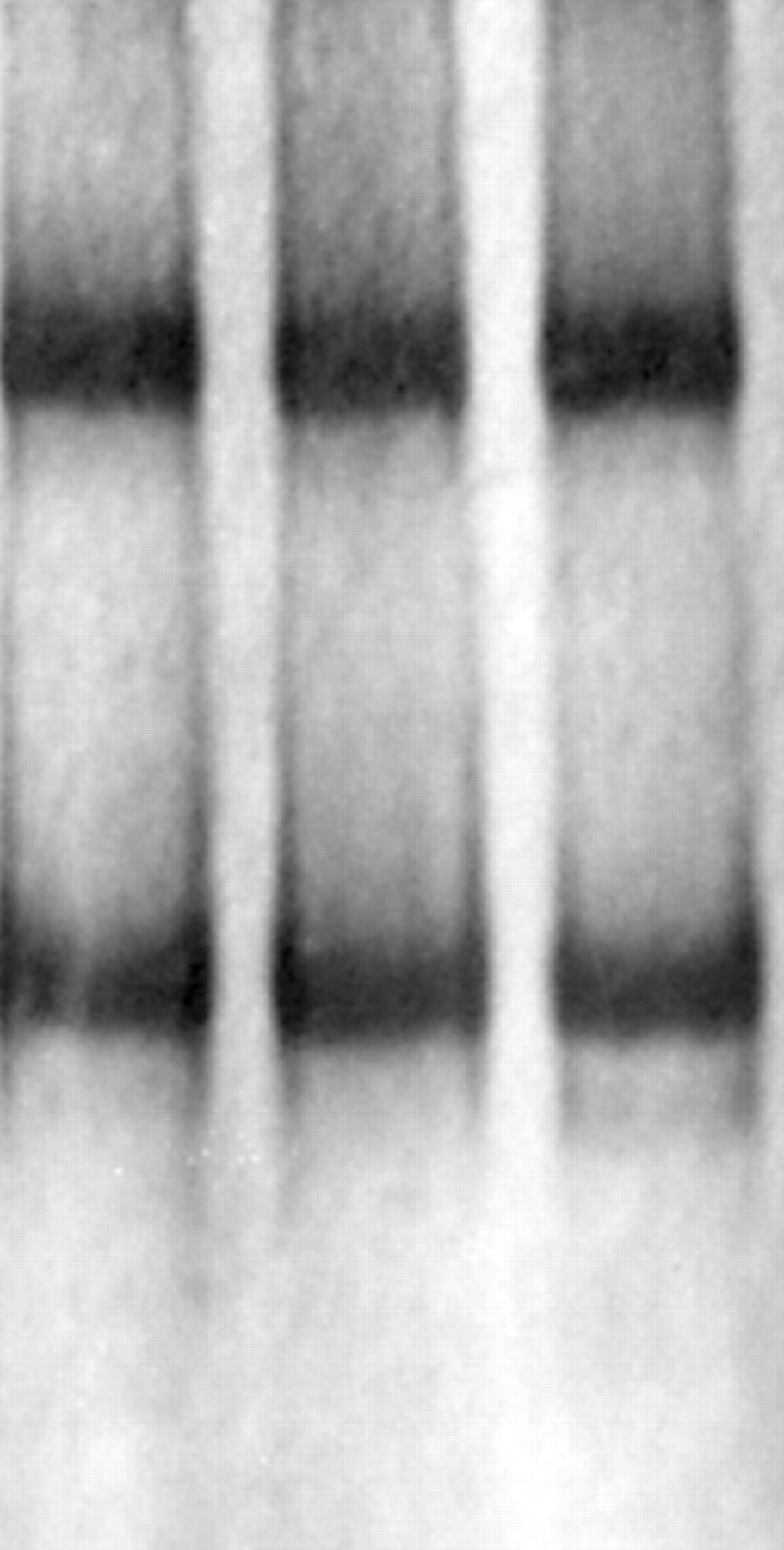
A band shift was identified by heteroduplex analysis in tumour tissue, but not in lymphocyte DNA, from patient T7 (fig 2). This somatic change, identified in a plexiform neurofibroma from patient T7, involved a CGA→TGA nonsense mutation at nucleotide 2446 of exon 16, resulting in the formation of a premature stop codon at residue 816 (fig 3A). The constitutional mutation in this family was a nucleotide substitution at the obligate donor GT dinucleotides (IVS4+1 G→A, intron 4), which was detected in the patient and two affected relatives (fig 3B). RNA was unavailable to confirm splice site abnormalities. A further, novel, somatic change (7168delGA) in exon 40 was detected in one of two neurofibromas obtained from the same patient (T52). Although the corresponding blood DNA was not available from this patient, the mutation was assumed to be somatic as it was not detectable in the second tumour, as would be expected if it represented a germline mutation.

Heteroduplex analysis of exon 16. The arrow points to extra band in patient T7.

{kind=link}
{kind=link}
{kind=link}
(A) Sequence analysis of the somatic alteration identified in patient T7, a nonsense mutation (R817X) at codon 2446 of exon 16. Lane (A) lymphocyte DNA from the patient, lane (B) DNA from patient's plexiform neurofibroma, lane (C) lymphocyte DNA from patient's mother. (B) Sequence analysis of germline mutation showing a donor splice site change in the obligate GT doublet (IVS4+1 G→A) observed in all affected members of the family of patient T7. Lane A is control DNA, lane B is patient T7 lymphocyte DNA, lane C is patient's tumour DNA, and lane D is lymphocyte DNA from patient's affected mother.
Discussion
Mutation analysis of the NF1 gene has so far identified constitutional mutations in 246 unrelated patients (NF1 Genetic Analysis Consortium, February 1999).30Approximately 70% of these disease causing sequence alterations are frameshift or nonsense mutations that are all predicted to result in the synthesis of a prematurely truncated neurofibromin protein. To date, despite the extensive investigation of theNF1 gene by a number of different mutation screening methods, disease causing lesions have been identified in less than 40% of patients.31 32 The spectrum ofNF1 germline mutations includes gross deletions, microdeletions, insertions, and base pair substitutions.31 The NF1somatic mutational spectrum is, however, far less well defined, mainly because of the small number of studies carried out and the difficulty in detecting such changes. This study aimed to investigate the possible mutational mechanisms involved in the somatic inactivation of theNF1 gene in various NF1 related tumour tissues.
Searches for evidence of loss of heterozygosity (LOH) of theNF1 gene region have proved successful in identifying potential NF1 deletion mutations in various tumour tissues.10-12 33 Three of these studies have provided evidence for at least some degree of LOH of theNF1 gene region in neurofibromas from NF1 patients. Coleman et al 10 found that 36% (8/22) of the neurofibromas analysed, derived from five unrelated NF1 patients, showed either partial or complete somatic deletions of one chromosome 17, and that these deletions always involved the NF1 region.10These results were essentially corroborated in a recent study which showed evidence of LOH in 25% (15/60) of neurofibromas tested from 17 NF1 patients; again the deletions always included theNF1 gene region.11 In contrast to these reports, showing clear evidence for LOH in more than a third of all neurofibromas analysed, is the recent study from Daschneret al 12 who found LOH in 2.6% of the 38 neurofibromas they investigated. Our study detected a higher level of LOH (12%) in neurofibromas than Daschneret al 12 but it is still significantly less than the previous reports.10 11 In none of the above studies were the constitutional mutations identified.
In the present study, DNA was isolated from 82 neurofibromas obtained from 45 unrelated NF1 patients, the largest panel studied to date. Following a screen with a large panel of NF1 intragenic and extragenic polymorphic markers, we identified LOH in 12% (10/82) of the tumour tissues tested. Indeed, in one case, 12 individual neurofibromas were tested from one NF1 patient and none showed any evidence of genomic deletions with any of the markers screened. To date, the constitutional mutations in six out of seven of the NF1 patients that have shown LOH in their tumours have still to be determined.
Some of the reasons for the low mutation detection rate could be because the lesions are too small to identify, the alterations being located in the regions still to be screened, or, possibly, the mutation detection techniques used are not sensitive enough. Thorough SSCP/HA analysis of NF1 exons 16-40 for sequence changes has, to date, only detected two somatic mutations and five novel germline mutations (three deletions and two splice site changes) (table 2). One of these alterations is located in the GRD region.
Neurofibromas are composed of a mixture of cell types (fibroblasts, mast cells, Schwann cells, perineurial cells) and it is not clear which cell types carry the genetic alterations underlying neurofibromas. Furthermore contamination of tumour DNA by normal cellular DNA will make LOH difficult to detect. Despite every effort to dissect any surrounding normal tissue carefully, it is always difficult to quantify the levels of contaminating normal cells present in any sample of dissected neurofibroma.
Ascertainment of the mutational events underlying the potential “second hit” leading to tumourigenesis in NF1 related tumours proved difficult, with only one study reporting the characterisation of both the germline and somatic changes of theNF1 gene in a dermal neurofibroma from a NF1 patient.34 The somatic nonsense mutation R816X detected in the plexiform neurofibroma from patient T7 in our study generates a premature protein at residue 816 and the same mutation was previously identified as a disease causing mutation in the lymphocyte DNA from an unrelated NF1 patient.36 The constitutional mutation in patient T7 identifies a splice site change IVS4+1 G→A, and this represents a novel mutation of the NF1 gene.
If we assume that our current mutational screening methodologies are sensitive enough to detect the majority of sequence alterations present, then these results would indicate that other mutational mechanisms affecting the NF1 gene are probably involved in neurofibroma formation, and other tumour suppressor genes may play an important role in NF1tumourigenesis. One inactivating mechanism being increasingly recognised in many tumour suppressor genes is the methylation of gene control regions. Hypermethylation of the gene promoter region has become an important alternative mechanism to specific coding region mutations for the inactivation of a number of tumour suppressor genes during neoplasia.36-39 We are currently examining the methylation status of the NF1 promoter region as a potential mechanism for gene silencing. Such hypermethylation of the NF1 gene may account for the low detection rate of somatic mutations within theNF1 coding region in NF1 associated tumours.
The determination of the underlying somatic mutational spectrum is important in helping us to understand the pathogenesis of theNF1 gene better. Before this current study, somatic alterations of the NF1 gene in a plexiform neurofibroma had only been indirectly shown by LOH studies. We are the first to characterise both the germline and somaticNF1 mutations from within such a tumour. Plexiform neurofibromas are of interest as they appear to be the site of malignant neurofibrosarcoma transformation and hence a better understanding of their developmental mechanism should provide us with further insight into the potential involvement of somatic mutations in NF1 tumourigenesis.
Acknowledgments
We would like to thank Professor David Cooper and Professor Peter Harper for their encouragement and Dr Nick Thomas for his comments on this manuscript. We are grateful to Dr Susan Huson and all other clinicians who have provided clinical material for this research and the patients and other families for their support. Financial support was provided by the Smiths Charity and the Neurofibromatosis Association.