Article Text

Abstract
The gene responsible for DNFB1 and DFNA3, connexin 26 (GJB2), was recently identified and more than 20 disease causing mutations have been reported so far. This paper presents mutation analysis for GJB2 in Japanese non-syndromic hearing loss patients compatible with recessive inheritance. It was confirmed that GJB2mutations are an important cause of hearing loss in this population, with three mutations, 235delC, Y136X, and R143W, especially frequent. Of these three mutations, 235delC was most prevalent at 73%. Surprisingly, the 35delG mutation, which is the most commonGJB2 mutation in white subjects, was not found in the present study. Our data indicated that specific combinations of GJB2 mutation exist in different populations.
- connexin 26
- GJB2
- Japanese
- non-syndromic hearing loss
Statistics from Altmetric.com
Connexin 26 gene (GJB2) mutations, first reported by Kelsell et al 1 in 1997, have recently been of particular interest because these mutations account for up to 50% of congenital deafness.2-4 To date, more than 20 disease causing mutations in GJB2 have been reported. One mutation, namely 35delG (also called 30delG), accounts for the majority of mutations detected.2-6 Since this mutation has been found across different populations, including those in the United States, France, Spain, Italy, the UK, Israel, New Zealand, Algeria, Pakistan, Morocco, and Tunisia,2 4-6 it is generally accepted that this six guanine residue sequence starting at position 30 is a hypermutable region. Since there have been no reports regardingGJB2 in Asian populations, it is an interesting question whether GJB2 mutations are also an important cause of non-syndromic recessive hearing loss in the Japanese population. We performed mutation screening forGJB2 in non-syndromic hearing loss families, including those with cases of sporadic deafness, which were compatible with recessive inheritance. We describe the unique spectrum ofGJB2 mutations found in the Japanese.
Subjects and methods
Thirty five families, in which one or two sibs were found to have bilateral sensorineural hearing loss and which were compatible with recessive inheritance of non-syndromic hearing loss, were used in this study. These included 10 families in each of which two sibs were affected and 25 families in each of which only one sib was affected (sporadic deafness cases). There were no syndromic features and the hearing of all parents was normal. All subjects had given informed consent for participation in the project. They were clinically well characterised by a series of auditory examinations (pure tone audiometry) and had sensorineural hearing loss, ranging from mild to profound in severity. All subjects underwent CT scans and none showed inner ear malformation.
MUTATION ANALYSIS
DNA fragments containing the entire coding region were amplified from genomic DNA samples using the primer pair Cx48U/Cx1040L (5′-GGTGAGGTTGTGTAAGAG TTGG-3′/3′-AGCAGAGCTCATTGTGGC ATC-5′) and used as a template for sequencing. An additional primer, Cx433U (5′-CT GCAGCTGATCTTCGTGTCC-3′), was also used for sequencing.
PCR conditions were as follows: five minutes denaturation at 95°C followed by 37 three step cycles (95°C for 30 seconds, 60°C for 30 seconds, 72°C for 2.5 minutes), followed by 72°C for 10 minutes and ending with a holding period at 4°C in a Perkin-Elmer thermal cycler. PCR products were directly sequenced after removing unincorporated dNTPs and primers by incubation at 37°C for 30 minutes with 50-100 ng PCR products with 0.1 μl exonuclease I (Amersham Life Science Inc, Cleveland, USA) and 1 μl shrimp alkaline phosphatase (Amersham Life Science Inc, Cleveland, USA). The enzymes were heat inactivated at 80°C for 15 minutes. Four pmol of either primer Cx48U or Cx433U were used in standard cycle sequencing reactions using ABI BigDye terminators and run on an ABI 377 sequencer. DNA samples from 96 unrelated Japanese, who had normal hearing, were used as controls.
Results
Table 1 summarises the mutations observed in this study. Analysis of GJB2 showed seven mutations, including five novel mutations, which segregated with the hearing loss phenotype, in a total of 13 affected subjects. Three missense mutations, a nonsense mutation, and three frameshift mutations were detected. Three common sequence changes, which may be non-pathological polymorphic changes, were also found. Fig 1 shows the pedigrees and sequence summaries for families with GJB2 mutations. These mutations were found in two (out of 10) families with two affected sibs each and in nine (out of 25) families with sporadic deafness cases. Families 1 and 2 were homozygous for a 235delC mutation. Eight families (3-10) were compound heterozygotes for (1) 235delC/Y136X, (2) 235delC/R143W, (3) R143W/176-191del16, (4) 299-300 ATdel/G45E, or (5) V37I/R143W. Family 11 was heterozygous for 235delC, but with no other mutations detectable (fig 1). All families including those with sporadic deafness cases were compatible with recessive inheritance (fig 1). A minus sign indicates the mutated allele and a plus sign indicates the normal sequence.
GJB2 mutations in Japanese
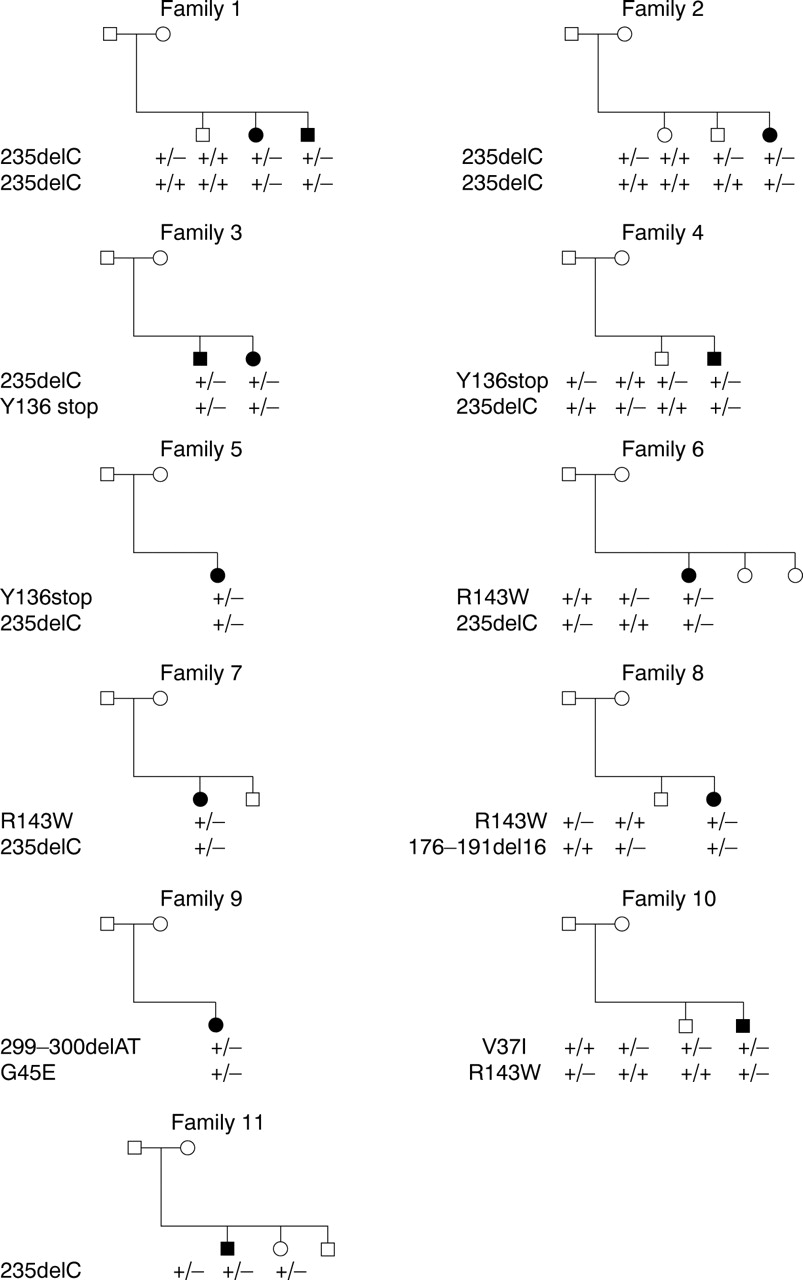
{kind=link}
Pedigrees of affected families and individual sequence summaries.
MISSENSE MUTATIONS
Three missense mutations, R143W (427C→T), G45E (134G→A), and V37I (109G→A), were detected. R143W was found in four families (Nos 6-8, 10). The patients bearing the mutations are compound heterozygotes (fig 1). G45E and V37I were also found to be compound heterozygous with other mutations in families 9 and 10 (fig 1). Two control subjects (out of 192 alleles) had a V37I mutation. The other two mutations were not detected in any of the 96 normal controls (192 alleles).
NONSENSE MUTATIONS
Three families (3-5) had a Y136X (408C→A) mutation, a C to A transition at residue 408. This nonsense mutation converts a tyrosine residue (TAC) at codon 136 to a stop codon (TAA). This mutation was not found in any of the 96 normal controls (192 alleles).
DELETION/FRAMESHIFT MUTATIONS
Three deletion frameshift mutations were detected. The 235delC mutation, which causes a frameshift at codon 79 resulting in a truncated polypeptide, was found in homozygosity in two families (1, 2), and compound heterozygous with the other mutations in five families (3-7). One family was heterozygous for 235delC, but with no other mutations detectable (fig 1). This deletion was found in two of the 96 controls (192 alleles). The second type of deletion, 176-191del16, was found in family 8. This deletion causes a frameshift leading to an altered amino acid sequence from codon 59, followed by a stop at codon 76. The other deletion, 299-300delAT, which causes a frameshift leading to an altered amino acid sequence from codon 100, followed by a stop at codon 113, was found in family 9.
MUTATIONS FOUND IN UNAFFECTED CONTROLS
GJB2 genotypes were analysed by direct sequencing for 96 unrelated subjects known to not have noticeable hearing loss (table 1). Three common sequence changes, V27I (79G→A), E114G (341A→G), and I203T (608T→C), were frequently found in control samples. The allele frequency was 39% (75/192), 13% (25/192), and 8.3% (16/192) respectively. Two subjects had the V37I missense mutation and the deletion frameshift mutation 235delC, described above (allele frequency 2/192=1.04%). The other mutations identified in affected families were not detected in the control samples.
Discussion
Approximately one third of affected families compatible with recessive inheritance, including those with sporadic cases, can be accounted for by GJB2 mutations. Therefore, the present results indicated that mutations inGJB2 are, as in other ethnic groups, an important contributor to recessively inherited non-syndromic hearing loss in Japanese ethnic groups.
With regard to the mutation types, the present findings showed that the spectrum of polymorphisms/mutations in GJB2in Japanese is significantly different from that found in ethnic groups with European ancestry. Seven mutations, of which five are novel, were identified in Japanese recessive families. Since five out of seven mutations represented here were not found in any of the controls, it is likely that these are potentially pathological mutations rather than non-pathological polymorphic changes. Although 235delC and V37I mutations were also detectable in two of 96 control subjects (192 alleles), frequent cosegregation with affected subjects indicates that they may be disease causing mutations. The 1.04% frequency (2/192 alleles) is compatible with the commonly found mutated allele frequency. Three common polymorphisms, V27I, E114G, and I203T, were found in control samples. There was no correlation with hearing loss, suggesting that these are non-pathological polymorphic changes. One of them, V27I, has already been reported as polymorphic (1/192 alleles),4 but it is noted that the frequency is extremely high in Japanese. The high frequency of the other two polymorphisms, which had not been previously reported, was also common in Japanese. Further study is needed to analyse the extent of this singularity.
This study also showed a high prevalence of three of these mutations, 235delC, Y136X, and R143W. Among them, 235delC was the most frequent and was found in eight of the 11 affected families (73%). Of the two mutations previously reported, R143W was detected in African families.7 Although the V37I mutation has been reported as polymorphic,4 one family in this study bearing this mutation was found to be compound heterozygous with R143W. Surprisingly, no subjects in this study had the 35delG (30delG) mutation, known to account for a majority of mutant alleles in white subjects.2 4 5 This mutation occurs in a stretch of six G residues at positions 30-35 of the GJB2DNA sequence and was detected in 28% (33/116 mutant alleles) of white US families of northern and southern European origin,4 and in 63-85% of mutant alleles in subjects with European ancestry.2-6 The negative data for 35delG in the Japanese population and the absence of 235delC in white patients suggests a founder effect for deafness in each population group. Actually, recent studies showed unique types of GJB2mutations in different ethnic groups, such as the 167delT mutation in Ashkenazi Jews4 8 and the R143W mutation in several families in a Ghanaian village.7 Haplotyping of the families with the 235delC mutation, the most prevalent mutation in Japanese, needs to be performed in order to evaluate the hypotheses relating to the origin of this mutation.
In conclusion, future screening programmes must take into account differences in these mutations among different ethnic groups. Particularly, it should be emphasised that 35delG may not be a common mutation in non-European ethnic groups.
Acknowledgments
These results were presented at the 22th ARO Meeting on 14-18 February 1999 at St Petersburg Beach, FL, USA (see the connexin 26 homepage, http://www.iro.es/cx26deaf.html). We thank all the families who participated in the present project. We would also like to thank Y Kon, Tsuyoshi Sakai, and Kyokugen Loo for technical assistance, and A C Apple-Mathews for help in preparing the manuscript. This study was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science and Culture of Japan (SU and HS), the Ministry of Health and Welfare of Japan (SU), and by NIH-NIDCD PO1 DC01813 (WJK).