Article Text

Abstract
The frequency, origin, and phenotypic expression of a germline MSH2 gene mutation previously identified in seven kindreds with hereditary non-polyposis cancer syndrome (HNPCC) was investigated. The mutation (A→T at nt943+3) disrupts the 3′ splice site of exon 5 leading to the deletion of this exon from MSH2 mRNA and represents the only frequent MSH2 mutation so far reported. Although this mutation was initially detected in four of 33 colorectal cancer families analysed from eastern England, more extensive analysis has reduced the frequency to four of 52 (8%) English HNPCC kindreds analysed. In contrast, the MSH2 mutation was identified in 10 of 20 (50%) separately identified colorectal families from Newfoundland. To investigate the origin of this mutation in colorectal cancer families from England (n=4), Newfoundland (n=10), and the United States (n=3), haplotype analysis using microsatellite markers linked to MSH2 was performed. Within the English and US families there was little evidence for a recent common origin of the MSH2 splice site mutation in most families. In contrast, a common haplotype was identified at the two flanking markers (CA5 and D2S288) in eight of the Newfoundland families. These findings suggested a founder effect within Newfoundland similar to that reported by others for two MLH1 mutations in Finnish HNPCC families. We calculated age related risks of all, colorectal, endometrial, and ovarian cancers in nt943+3 A→T MSH2 mutation carriers (n=76) for all patients and for men and women separately. For both sexes combined, the penetrances at age 60 years for all cancers and for colorectal cancer were 0.86 and 0.57, respectively. The risk of colorectal cancer was significantly higher (p<0.01) in males than females (0.63v 0.30 and 0.84 v0.44 at ages 50 and 60 years, respectively). For females there was a high risk of endometrial cancer (0.5 at age 60 years) and premenopausal ovarian cancer (0.2 at 50 years). These intersex differences in colorectal cancer risks have implications for screening programmes and for attempts to identify colorectal cancer susceptibility modifiers.
- MSH2 mutation
- HNPCC
- colorectal cancer
Statistics from Altmetric.com
Hereditary non-polyposis colorectal cancer syndrome (HNPCC) is characterised by a dominantly inherited susceptibility to the development of early onset colorectal cancer, although with age dependent but incomplete penetrance. The syndrome can be clinically classified according to the presence or absence of extracolonic cancers (Lynch II and Lynch I, respectively). These extracolonic cancers typically occur in the uterus, ovaries, urogenital, and upper gastrointestinal tracts.1-3 Recently germline mutations in DNA mismatch repair (MMR) genes, including MSH2, MLH1, PMS1, and PMS2, have been identified in HNPCC families,4-8 with alterations to MSH2 and MLH1 accounting for approximately 90% of classically affected kindreds.9-12 There appears to be no clear correlation between genotype (MSH2 or MLH1) and phenotype, except for a suggestion that extracolonic cancers may be more common in MSH2 kindreds.13
The mutation spectrum in HNPCC is wide, with mutations identified along the entire length of the coding sequence in both major genes, although in MLH1 exons 15 and 16 may be more frequently affected than other regions.14 The majority of mutations reported so far are protein truncating, but missense mutations and large deletions also occur.10 12 15 16 The maximal reported detection rate of MMR mutations in strictly defined (“Amsterdam criteria”) kindreds is approximately 70%, even by high input, direct sequencing of coding regions.10 18 Therefore, mutation detection screening services offered to HNPCC families may involve lengthy, labour intensive analyses. Linkage analysis is often not feasible owing to the size or structure of the presenting family. In Finland, haplotype analysis has shown founder effects (common genealogical ancestor) for two distinct mutations, in approximately 63% of HNPCC kindreds, greatly simplifying family screening.18 19However, in the UK and USA very few recurrent mutations have been identified, and the relative frequencies of protein truncating and missense mutations are as yet unclear.
To date, the single most common MSH2 mutation is a point mutation, A→T at nucleotide 943+3 (A→T nt943+3) in the 3′ splice site of exon 5, which results in the deletion of exon 5 in mRNA and a truncated protein. Originally identified in three of 29 North American HNPCC families,10 it was subsequently detected in four of 33 English kindreds,20 with a predicted incidence in England of about 12%. We have investigated the origin of the A→T nt943+3 MSH2 mutation to determine if there is evidence of a founder effect in England and North America as reported for MLH1 mutations in Finland.18 19 In addition, the penetrance and phenotypic expression of the mutation was determined in 76 gene carriers.
Material and methods
PATIENTS
Seven families with the A→T at nt943+3 mutation were originally ascertained through referral to cancer family clinics in the UK and in North America. One family (family C) from Newfoundland is particularly extensive (it was originally ascertained as two apparently unrelated kindreds) and was used in the original study showing linkage to 2p.21 Colorectal cancer families from England and Newfoundland were screened for evidence of the A→T nt943+3 mutation as described below. All families studied satisfied modified Amsterdam criteria such that ovarian or endometrial cancer was allowed as an alternative to a single colorectal cancer in the standard criteria. For mutation positive kindreds, information on age at onset and type of cancers in gene carriers was collected from family members and confirmed by hospital records or death certificates whenever possible. Subjects were considered to be affected if (1) they were shown to be a mutation carrier or (2) if they were clinically affected (colorectal, uterine, or ovarian cancer) and a first degree relative was shown to be a mutation carrier.
MOLECULAR GENETIC ANALYSIS
The presence of the exon 5 3′ splice site point mutation (A→T at nt943+3) was originally detected by in vitro coupled transcription/translation (IVTT), sequencing, and restriction digestion.10 20 For this study, all subjects were reanalysed for the presence of the mutation by single stranded conformational polymorphism (SSCP) as follows: 5/8 UTaq polymerase (Life Technologies) was used to amplify MSH2 exon 5 and flanking intronic sequence from 250 ng genomic DNA with primers 5F (5′ GTG GCT ATA GGA AAT CTT CGA 3′) and 5R (5′ ACC ATT CAA CAT TTT TAA CCC 3′) in 2.5 mmol/l MgCl2 PCR buffer. After confirming successful amplification of the 246 bp product by agarose gel electrophoresis, samples were denatured and loaded onto precooled (5°C) 0.5 × MDE (mutation detection enhancement gel, J T Baker), 5% glycerol, 0.5 × TBE gels for single strand conformational polymorphism analysis (SSCP). Gels were run for a total of 80 W hours, and then silver stained, as follows: after fixing in 10% ethanol, 0.5% acetic acid for 10 minutes, the gel was incubated for 15-20 minutes in 0.1% aqueous silver nitrate. The gel was then rinsed briefly with distilled water and developing solution (1.5% NaOH, 0.2% formaldehyde) added. When suitably strong product colouration was obtained, the developer was poured off and the gel left to fix in 0.75% sodium carbonate for 20-60 minutes, after which photographic records were made.
PCR samples showing the aberrant SSCP pattern were purified (Wizard Purification kit, Promega) and sequenced using primer 5F (γ32P end labelled with T4 polynucleotide kinase) andfmol cycle sequencing kit (Promega). Products were electrophoresed at 60 W for four hours on a 60 cm 6% denaturing polyacrylamide gel and the autoradiographs developed after overnight exposure. All samples with the aberrant SSCP pattern possessed the A→T at nt943+3 mutation.
During the study, affected members of 11 additional families originating from the same isolated geographical region of Newfoundland as family C and from eight families from the rest of Newfoundland were tested for the A→T at nt943+3 mutation by SSCP and sequencing (fig1). Mutation carriers identified were then included in the haplotyping studies.

SSCP gel analysis of large Newfoundland kindred. Numbering lanes from left to right: lanes 1 and 18 size markers, lanes 2 to 17 DNA samples. Lanes 2, 3, 9, 11, 12, 14, 15, and 17 show abnormal band shifts and were found to be nt943+3 A→T MSH2 mutation carriers.
Haplotype analysis was performed using five microsatellite markers flanking MSH2: D2S391, D2S288, CA5, D2S123, and D2S378.5 22 One of each primer pair was γ32P ATP end labelled with T4 polynucleotide kinase and included in the PCR, which was performed under previously optimised conditions.20 PCR products were electrophoresed on 6% denaturing polyacrylamide gels. Alleles were sized by comparison with alleles of affected members from all families run on the same gel, and by comparison to a radiolabelled sequencing ladder. Haplotypes were then constructed and compared.
To determine the frequency of the haplotypes identified in the UK kindreds, the microsatellites were tested against a panel of 20 parent/child pairs from kindreds with a predisposition to colorectal cancer (either APC or HNPCC). For each pair, previous studies had shown that neither subject carried the causal germline mutation in that family, thus these were essentially normal UK haplotypes.
It was not possible to ascertain a similar panel of samples from Newfoundland. Instead, 16 unrelated spouses from Newfoundland HNPCC and von Hippel-Lindau disease families were tested with this panel of microsatellites and allele frequencies obtained for comparison with the mutation carrying kindreds identified in this study.
STATISTICAL ANALYSIS
Age related cancer risks were calculated by life table analysis using the age to first cancer. Intersex differences in cancer risks were compared using the log rank test.23 Statistical significance was taken at the 5% level.
Results
FREQUENCY OF THE MSH2 (NT943+3 A→T) MUTATION AND HAPLOTYPE ANALYSIS
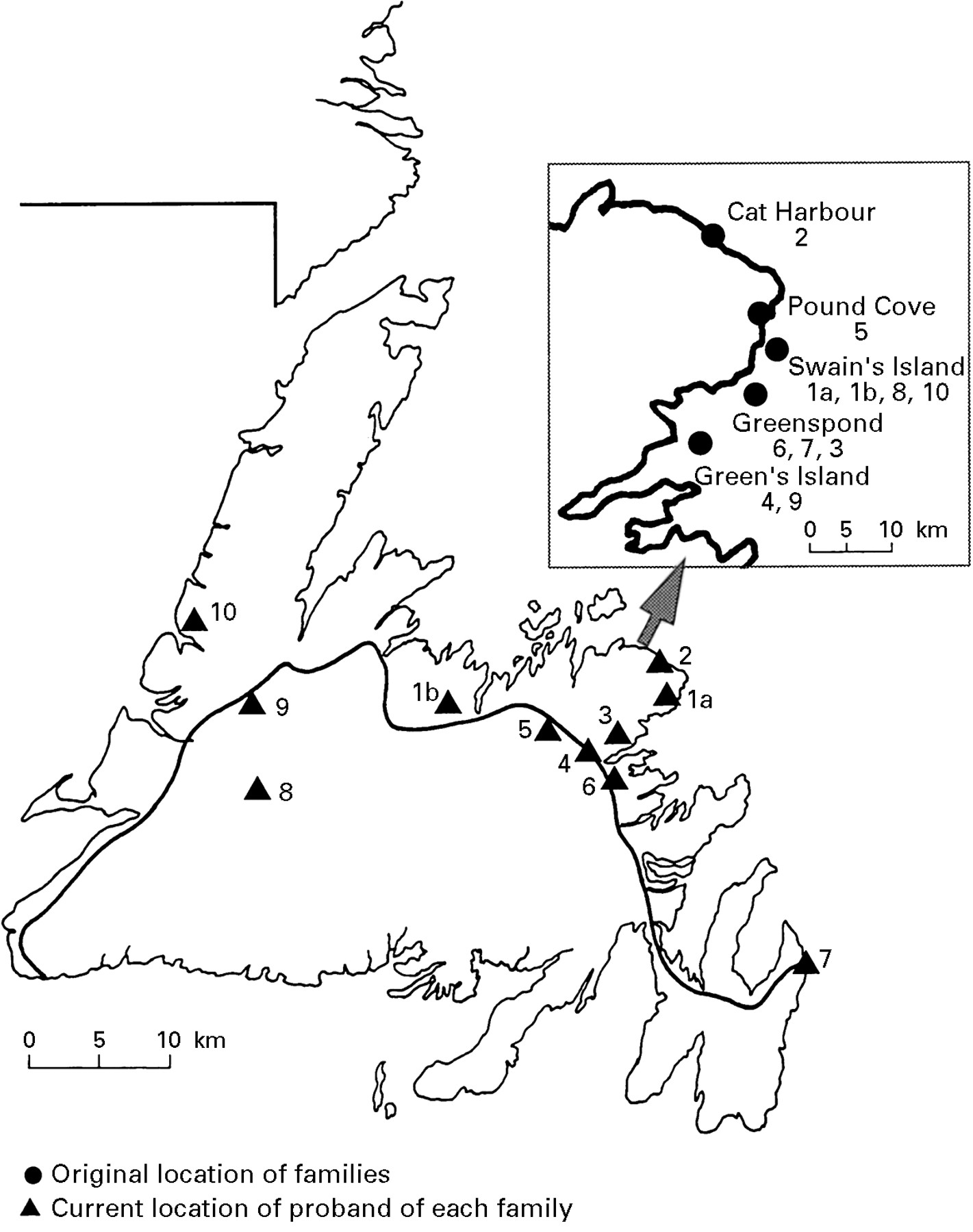
A total of 52 probands from familial colorectal cancer kindreds from eastern England were analysed for the MSH2 exon 5 splice site mutation. Apart from the four families reported previously (out of 33 tested),20 no further families with this mutation were identified (in a further 19 samples), providing an overall incidence of 8% (4/52) of all HNPCC families tested. The large Newfoundland HNPCC kindred (family C) reported previously21 originated in the Bonavista Bay region of north east Newfoundland (fig 2). To determine the incidence of the nt943+3 A→T MSH2 mutation in Newfoundland, a further 19 kindreds with familial colorectal cancer were analysed and the MSH2 mutation was identified in nine families. Through extensive archival research, ancestors of all nine families were shown to have originated from the north eastern coastal region close to the origin of family C (fig 2). However, none of these families could be connected by a common ancestor.

{kind=link}
{kind=link}
Map of Newfoundland showing original and current location of proband of 10 Newfoundland families with nt943+3 A→T mutation. The insert shows a large scale map of the Bonavista Bay area.
To investigate the origin of the nt943+3 A→T MSH2 mutation in different kindreds, haplotype analysis was performed in the 10 Newfoundland families, four English families, and three families from the United States with microsatellite markers linked to MSH2 (table 1). Eight of 10 Newfoundland families displayed a common haplotype over a minimum 5.9 cM region containing D2S391, D2S288, MSH2, CA5, and D2S123. Genotyping of 16 unrelated controls from Newfoundland showed that none carried the haplotype identified in the Newfoundland nt943+3 A→T MSH2 mutation families (data not shown). Two Newfoundland families (N2 and N10) had a different haplotype at two markers (D2S391 and D2S288) distal to MSH2 from the other eight families, but had an identical haplotype at three centromeric markers (CA5, D2S123, and D2S378) contained within a 9.2 cM interval. Although it may be that families N2 and N10 have a common origin with the other Newfoundland families, but are descended from a person in whom there has been a recombination between D2S288 and MSH2, this cannot be proven and N2 and N10 showed different alleles at D2S391.
Haplotypes of North American and UK kindreds. The marker order is tel-D2S391-0 cM-D2S288-2.1 cM-CA5-3.8 cM-D2S123-5.4 cM- D2S378-cen. MSH2 maps between CA5 and D2S288
For the three families from the United States, there was no unequivocal evidence of an extended haplotype shared with the Newfoundland families, although the CA5 allele identified in the Newfoundland families was present in two of three US families. This allele was the most common allele found in controls and accounted for 36% of CA5 alleles detected. An unequivocal haplotype could be established for three of the four English families tested, and there was no evidence of a single common haplotype in the English families. Although the ancestors of family C were known to have emigrated from England in ∼1750, the allelotype of the majority of Newfoundland families differed at the D2S288 locus from each of the English families and at CA5 from three of the four English families. Hence, there was no clear evidence of a common origin for the English and Newfoundland families.
PENETRANCE AND PHENOTYPIC EXPRESSION
Subjects from the four English HNPCC families and the Newfoundland families were tested to determine their carrier status; 76 gene carriers were identified and 45 of these had developed cancer (27 females and 18 males). The most common sites of cancer were colorectal (n=29), endometrial (n=15), and ovarian (n=8). Infrequent sites included ureter and bladder (n=3), skin (n=3), stomach (n=2), and there was one case each of breast, duodenal, jejunal, and kidney cancer. The mean (SD) age at diagnosis of first cancer was 42.9 (10.1) years and the mean age of diagnosis of colorectal cancer was similar (43 (10.5) years). However, the mean age at diagnosis of colorectal cancer was younger in males than in females (38.8 (7.9)v 47.2 (11.1), p<0.05). The age related risks for all cancers, colorectal, endometrial, and ovarian cancers were calculated by life table analysis (table 2). These showed that the risk of colorectal cancer in male gene carriers was significantly higher than in female heterozygotes (χ2=7.29, p<0.01). However, for female there were additional significant risks of endometrial and ovarian cancer, so that the overall cancer risks for the two sexes did not differ significantly (χ2=3.06, p>0.05).
Age related risks to first event for all cancers and for the principal site specific cancers (colorectal, endometrium, and ovary) in 76 subjects with a nt943+3 A→T MSH2 mutation. The risk of colorectal cancer in males was significantly higher than that in females (p<0.01), but the overall risk for all cancers did not differ between the two sexes
Discussion
Investigations of kindreds sharing a common mutation may provide new insights into the origin of genetic disease and the determinants of phenotypic expression of disease alleles. Although most HNPCC gene mutations are private, in Finland two MLH1 mutations have been reported to account for 63% of HNPCC families. These two mutations were shown to be derived from two ancestral chromosomes with affected families sharing the same haplotype.18 19 Although some of our families with the common MSH2 mutation shared an identical haplotype, there was no evidence of a common origin for all families. Thus, of the seven English and US families studied, most showed distinct haplotypes and there was no evidence of a common haplotype at flanking markers. Similarly, the Newfoundland family C, which is known to have originated in south western England and travelled to Newfoundland in 1757, did not appear to be closely related to any of the English families. Hence these results do not provide evidence of a recent founder effect to explain the world wide frequency of the exon 5 splice site MSH2 mutation. However, it is difficult to exclude with certainty an ancestral founder effect with divergence of disease haplotypes. Although microsatellite markers are frequently used for haplotype analysis because of their informativeness, an ancestral mutation could exist on different haplotypes because of recombination between the mutation and the linked markers or because of de novo mutations in the number of dinucleotide repeats in microsatellite loci, or both. This is known to occur at a low but finite frequency in normal families. Although there is no evidence for an increased mutation rate during meiosis in subjects with germline MSH2 or MLH1 mutations, a small effect cannot be excluded. Nevertheless, microsatellite markers were used successfully to show two common haplotypes in apparently unrelated Finnish HNPCC families with MLH1 mutations believed to date from the 16th and 18th centuries.24 Thus, for the oldest Finnish MLH1 mutation, most families shared a conserved haplotype at markers spanning 2.0-15.0 cM around MLH1. Hence, the most likely interpretation of our data is that the world wide distribution of the nt943+3 A→T MSH2 mutation does not result from a single founder mutation. This conclusion is supported by reports of this mutation in HNPCC kindreds from Japan and Italy.25 26
Within Newfoundland there was clear evidence of a founder effect in most families. Following the discovery of Newfoundland in 1497, the first permanent settlement was established in 1610 but by 1750 the population of the whole island was only 6000. The peak years of immigration were from 1800-1830 when English Protestant settlers from the port cities of south west England (Devon and Dorset) settled in the eastern and north eastern bays (Conception, Bonavista, and Notre Dame) and along the south coast. Irish Catholic settlers populated the Avalon peninsula and other areas. Fishing was the primary industry and the majority of communities were coastal. Descendants of original settlers often clustered in nearby coves and offshore islands. Until recent generations, large family size was common and a series of genetic isolates with founder effects have been described within the Newfoundland population. The large family C was originally ascertained as two HNPCC families from the north eastern coastal region and (by extended pedigree studies and archival searches) shown to be connected by a single ancestor five generations previously. All nine of the additional families with the nt943+3 A→T MSH2 mutation could trace their origin to Bonavista Bay, and the haplotype data are also compatible with a common origin in at least eight of the Newfoundland families. The spread of the nt943+3 A→T MSH2 mutation in Newfoundland is consistent with that expected from population migration as a result of socioeconomic factors. Our finding of a MSH2 founder effect within Newfoundland is similar to that of MLH1 founder effects in Finland.
The precise comparison of cancer risk estimates in MSH2 and MLH1 mutation carriers is hindered by differences in methods of ascertainment, geographical variations in environmental factors, possible genetic modifiers, and allelic heterogeneity. The latter variable can be controlled for by analysing subjects with identical mutations. An important finding in our analysis is the higher risk of colorectal cancer in male gene carriers than in females. Although previous studies without confirmation of germline mutations had shown similar risks of colorectal cancer in both sexes,3 27 our findings confirm a recent report of a sex related difference in colorectal cancer risks by Dunlop et al,28 who studied MMR gene mutation carriers ascertained through a population based strategy. In addition, a study of proven MMR gene mutation HNPCC gene carriers also found a lower colorectal cancer risk in females, although the difference did not reach statistical significance.13 As these reports included a variety of MMR gene mutations, it seems unlikely that the observed sex differences in colorectal penetrance are specific to the nt943+3 A→T MSH2 mutation (only one of the six families analysed by Dunlop et al 28 had an exon 5 MSH2 mRNA deletion and this was caused by a different MSH2 mutation). The absolute risks of colorectal cancer in our cases were modestly raised over those calculated by Dunlop et al,28 reflecting the differences in methods of ascertainment. Similarly the endometrial cancer risk in our study was higher than that reported by Dunlop et al 28 (50% v 42% at age 70 years); however, in both studies the risk of endometrial cancer in female MMR gene mutation carriers was higher than that of colorectal cancer. Vasen et al 13 reported lifetime risks of 61% and 42% for endometrial cancer in MSH2 and MLH1 carriers, respectively. Estimates of endometrial cancer risk calculated from studies using molecular genetic carrier testing have provided higher risks than earlier studies.29 In our study, there was also a significant risk of premenopausal ovarian cancer (20%) in gene carriers. Although ovarian cancer is a well recognised feature of HNPCC, Vasen et al 13 found only a sixfold increase in relative risk of ovarian cancer in HNPCC gene carriers and Aarnio et al 27reported a 9% lifetime risk of ovarian cancer but a 19% risk of stomach cancer. Further studies are required to determine if the nt943+3 A→T MSH2 mutation is specifically associated with a higher risk of ovarian cancer than other MSH2 or MLH1 mutations. However, our data may influence female MMR gene mutation carriers in our HNPCC families towards prophylactic oophorectomy and hysterectomy after completing their family. To date, no clear correlation between HNPCC allelic heterogeneity and ovarian and uterine cancer risk has been reported, although the risk of extracolonic cancers is suggested to be higher with MSH2 mutations. In addition to allelic heterogeneity, cancer risks may be influenced by modifying effects such as rare ras alleles (as reported for ovarian cancer risk in BRCA1 kindreds30). The investigation of a large series of patients with a restricted spectrum of germline mutations would facilitate the identification of genetic modifiers and the factors determining the intersex differences in colorectal cancer risks.
Note added in proof
Since submission of this paper, another 21 HNPCC families have been identified in Newfoundland, and one of these (with ancestors from the same geographical region) has the same mutation (A→T at nt943+3).The proportion of families with this mutation is now 11 of 41 (27%). A common ancestor has now been identified for families 1 (N1), 8 (N6), and 10 (N2). A common ancestor has also been identified for families 4 (N8) and 9 (N7) (figs 1 and 2).
Acknowledgments
The first two authors contributed equally to this work. We would like to acknowledge the financial support of the Cancer Research Campaign, Addenbrooke’s NHS Trust, and the Canadian Genetic Diseases Network. We are grateful to all those clinicians who have provided samples.