Article Text

Abstract
Fanconi anaemia (FA) is an autosomal recessive disease characterised by congenital abnormalities, defective haemopoiesis, and a high risk of developing acute myeloid leukaemia and certain solid tumours. Chromosomal instability, especially on exposure to alkylating agents, may be shown in affected subjects and is the basis for a diagnostic test. FA can be caused by mutations in at least seven different genes. Interaction pathways have been established, both between the FA proteins and other proteins involved in DNA damage repair, such as ATM, BRCA1 and BRCA2, thereby providing a link with other disorders in which defective DNA damage repair is a feature. This review summarises the clinical features of FA and the natural history of the disease, discusses diagnosis and management, and puts the recent molecular advances into the context of the cellular and clinical FA phenotype.
- DNA repair
- Fanconi anaemia
- chromosome instability
- clinical management
Statistics from Altmetric.com
Fanconi anaemia (FA) is an autosomal recessive condition first described in 1927 by the Swiss paediatrician Guido Fanconi,1 who described a familial form of aplastic anaemia in three brothers with short stature, hypogonadism and skin pigmentation. Since then, over 900 cases have been reported2 enabling the disease to be clearly defined. FA cells are characterised by chromosomal hypersensitivity to cross linking agents3 and the resulting increase in chromosome breakage provides the basis for a diagnostic test.4 Complementation analysis by cell fusion and correction of cross linker hypersensitivity has delineated at least eight complementation groups (FA-A, B, C, D1, D2, E, F, G),5 six genes have been cloned (FANCA, C, D2, E, F, G),6–12 and a recent report has shown that FANCD1 and possibly also FANCB are actually BRCA2.13 As a result of these findings, the study of FA protein interactions has become possible and functional pathways between the FA proteins and other proteins involved in DNA repair have been established.14–16 This review will summarise the clinical features of FA, discuss diagnosis and management, and explain how the recent molecular advances have helped us understand the cellular and clinical phenotype.
EPIDEMIOLOGY
The incidence of FA is approximately three per million and the heterozygote frequency is estimated at 1 in 300 in Europe and the United States.17,18 FA has been reported in many ethnic groups19 and founder mutations have been described in Ashkenazi Jews, who have an approximate carrier frequency of 1 in 89,20 and Afrikaners where the carrier frequency was estimated at 1 in 83.21
CLINICAL FEATURES
Congenital abnormalities
FA is a very heterogeneous condition clinically and patients can have a wide variety of abnormalities (table 1).22,23
Frequency of abnormalities in FA (taken from Dokal, 2000131)
Of the skeletal abnormalities, radial ray defects such as hypoplasia of the thumbs and radial hypoplasia are the most common (figs 1A, 2A, B); other skeletal defects which may occur include congenital hip dislocation, scoliosis, and vertebral anomalies. Affected subjects often have generalised skin hyperpigmentation, café au lait spots, and areas of hypopigmentation (fig 1B). FA is associated with altered growth both in utero and postnatally. Low birth weight is common and the median height of FA patients lies around the 5th centile. This can sometimes be related to growth hormone deficiency or hypothyroidism; in a recent prospective study of 54 FA patients, 44% had a subnormal response to growth hormone stimulation and 36% had overt or compensated hypothyroidism.24 Abnormalities of glucose/insulin levels were also common and the authors concluded that while short stature is an integral feature of FA, superimposed endocrinopathies might further impair growth. Microphthalmia, microcephaly, and developmental delay are all features frequently present. Conductive deafness is also relatively common; this can be mild and may or may not be associated with external ear malformations. Renal abnormalities are present in approximately one third of patients and include unilateral renal aplasia, renal hypoplasia, horseshoe kidneys, or double ureters. In males there is a high incidence of genital abnormalities such as hypogenitalia, undescended testes, and hypospadias, and infertility is usual, although there have been reports of males with FA fathering children.25 Females may also have underdeveloped genitalia and uterine anomalies; older females have sparse, irregular menses but can become pregnant if not on androgen therapy.26

(A) Typical radial ray abnormalities and (B) café au lait patches and hypopigmentation, all common features in FA.
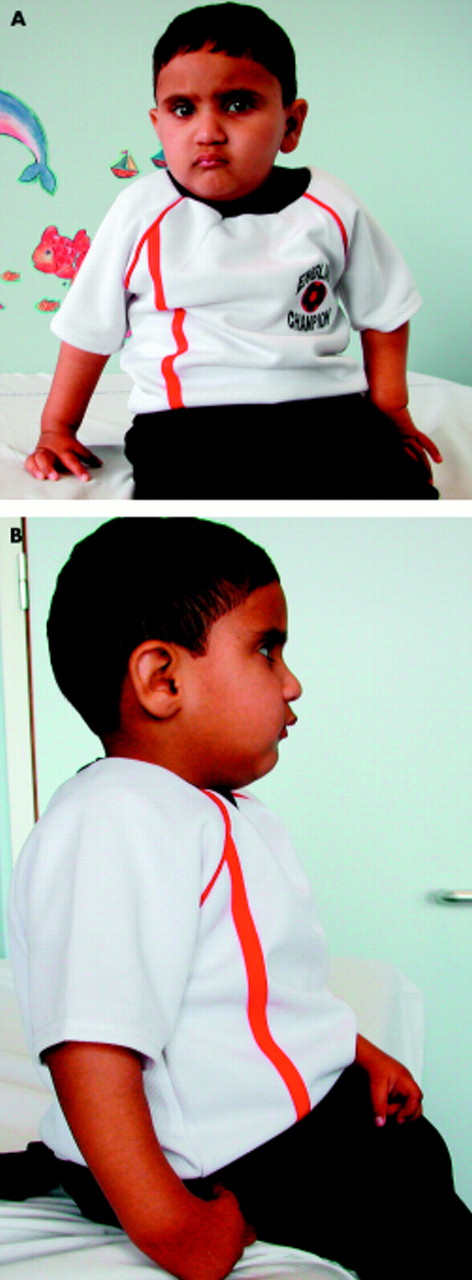
(A, B) A 3½12 year old FA child showing radial ray abnormalities. Height and head circumference are both below the 3rd centile.
Less common abnormalities in FA include gastrointestinal defects such as atresia (oesophageal, duodenal, jejunal), imperforate anus, tracheo-oesophageal fistulae, cardiac defects such as patent ductus arteriosus, ventricular septal defect, pulmonary stenosis, aortic stenosis, and coarctation, and central nervous system defects including hydrocephalus, absent septum pellucidum, and neural tube defects. Apart from isolated immune system abnormalities documented in case reports,27 there is no systematic evidence that FA patients have immune deficiencies before bone marrow failure.
There can be a large degree of phenotypic variability within families, as exemplified by Koc et al,28 who described four FA patients from two related consanguineous families who all had the same FANCA mutation but exhibited wide variation in birth weight, skin pigmentation, and the severity of skeletal, renal, and genital abnormalities. Importantly, up to one third of FA cases do not have any obvious congenital abnormalities, only being diagnosed when another sib is affected or when they develop haematological problems,29 although most do have some subtle clinical features such as dermatological manifestations. Phenotypes from each complementation group are broadly similar, discussed in more detail later.
Haematological abnormalities
The most important clinical features of FA are haematological; FA is the commonest type of inherited bone marrow failure syndrome and the incidences of aplastic anaemia, myelodysplastic syndrome (MDS), and acute myeloid leukaemia (AML) are all greatly increased in homozygotes. At birth, the blood count is usually normal and macrocytosis is often the first detected abnormality followed by thrombocytopenia and neutropenia. Pancytopenia typically presents between the ages of 5 and 10 years, the median age of onset being 7 years.30,31 Clinically, the affected FA patient may present with bleeding, pallor, and/or recurring infections. One study of 388 FA patients calculated the actuarial risk of developing haemopoietic abnormalities and death from haematological causes by the age of 40 to be 98% and 81%, respectively.31 The risk of pancytopenia was 84% by the age of 20 years, followed by clonal cytogenetic abnormalities (risk of 67% by 30 years) and MDS or AML (risk of 52% by 40 years). Of the patients studied, 135 (35%) died during the time of the study, the median age of death being 13 years, and the majority (120) died from haematological complications (49 from bone marrow failure, 37 from post transplant treatment related complications, and 34 from MDS or AML). Certain cytogenetic abnormalities are commonly seen in FA patients with MDS/AML, including monosomy 5 and monosomy 7.30 These clonal abnormalities are more often found in sporadic MDS related AML and AML occurring secondary to treatment with alkylating agents.32
In those affected patients who survive into early adulthood, there is a greatly increased risk of solid tumours, notably hepatic tumours, which may be related to androgen use, and squamous cell carcinomas of the oesophagus, oropharynx, and vulva.2
Studies of cancer risk in FA heterozygotes are conflicting; the initial study by Swift et al18 analysed 102 deaths in the relatives of eight FA cases and found a higher rate of leukaemia and cancers of the tongue, stomach, and large bowel. However, this was not statistically significant and when the study was expanded to 25 families, no overall or specific excess of cancers could be shown and there were fewer leukaemias than expected.33 A separate study of 125 relatives in nine FA families also failed to show a difference.34
In summary, FA is a highly heterogeneous syndrome; homozygotes can present at birth with congenital abnormalities or during childhood with haemopoietic abnormalities. The main causes of morbidity and mortality are aplastic anaemia, myelodysplasia, acute myeloid leukaemia, and solid tumours at older ages in those surviving the childhood haematological malignancies.
MANAGEMENT OF THE PATIENT WITH FA
Making the diagnosis
Because of the heterogeneous nature of the condition, FA is difficult to diagnose on clinical features. FA cells show increased chromosome breakage but this is highly variable and overlaps with the range for normal cells. Adding DNA cross linking agents such as mitomycin C (MMC)35 or diepoxybutane (DEB)36 increases the number of chromosome breaks with distinct ranges for normal and FA cells, so providing the basis for a diagnostic test.4 Chromosome breakage studies can be carried out on amniotic cells, chorion villus cells, or fetal blood and therefore it can be used for prenatal diagnosis.37 Although MMC/DEB testing is highly specific for FA, interpretation is complicated in cases of somatic mosaicism (see Somatic mosaicism section below).
FA patients have an intrinsic cell cycle disturbance consisting of prolonged progression through, and arrest within, the G2 phase compartment of the cell cycle38; flow cytometry of suspected FA patient cells treated with cross linkers can measure this and make the diagnosis with comparable accuracy to chromosome studies.39–41 This approach has the advantage that it is less time consuming and does not require cytogenetic expertise. It may also sometimes be helpful when there are discrepancies on DEB testing.42 However, it is not reliable in cases with concurrent myelodysplasia or leukaemia.40,41 Until recently, there has been no method to determine complementation group apart from time consuming cell fusion assays, but it has now been shown that retroviruses expressing FANCA, FANCC, or FANCG cDNA can be used to correct the phenotype of T cells from FA patients and thereby determine complementation group in a rapid, accurate manner.43
Differential diagnoses
Anomalies such as vertebral defects, tracheo-oesophageal atresia, and renal and radial ray defects found in the sporadic VATER/VACTERL association overlap with those found in FA.
Thrombocytopenia-absent radii (TAR) syndrome, which is autosomal recessive, presents with thrombocytopenia at birth or around the neonatal period and radial ray defects but, unlike FA, thumbs are invariably present bilaterally.44,45 Unlike FA, there is no documented increase in haematological or solid tumour malignancies in TAR.
Diamond-Blackfan anaemia is characterised by defective erythroid progenitor maturation and usually presents in the first year of life with normochromic or macrocytic anaemia.46 Over one third have congenital malformations, often involving the head (micrognathia, cleft lip), upper limbs, and genitourinary system. It is slightly more common than FA and most cases are sporadic with evidence of autosomal dominant or, less frequently, recessive inheritance.
In all three conditions, MMC/DEB testing is within normal parameters, allowing confident differentiation from FA.
Nijmegen breakage syndrome (NBS) is a rare autosomal recessive disorder resulting from mutations in NBS1, which codes for the nibrin protein.47 It is characterised by immune deficiency, microcephaly, and hypersensitivity to ionising radiation and the majority of patients are of Slav origin and are homozygous for a single mutation in the NBS gene (657del5), suggesting a founder effect.47,48 A recent study of NBS in eight Russian patients found three to have haemopoietic abnormalities or AML.49 NBS can therefore closely mimic FA at the clinical level, and testing for NBS mutations should be considered if there is any doubt regarding the diagnosis of FA.
Somatic mosaicism
In haemopoietic cells of FA patients, somatic mosaicism may result in the reversion from the pathogenic allele to the wild type, resulting in the disease phenotype reverting back to normal in the relevant cell clones; 10–25% of FA patients will have such mixed lymphocyte populations and this can create difficulty in interpreting diagnostic chromosome breakage tests. This is exemplified by a case of two affected brothers who had known FANCC mutations but only one a positive DEB test.50 Thus, somatic mosaicism could be one explanation for intrafamilial phenotypic variation in FA.
The exact mechanism for somatic mosaicism can vary; one mechanism involves mitotic recombination in a bone marrow stem cell, resulting in both FA mutations on the same allele while the other allele becomes functional.51 Other observed mechanisms include spontaneous genotypic reversion52 and functional correction of the mutation as a result of secondary sequence alterations in cis.53 All may confer a selective advantage to the cell, which then repopulates the marrow and could lead to improved haemopoiesis. While some FA somatic mosaics do have a milder haematological phenotype,51 this is not always the case.
Initial management
If a diagnosis of FA is suspected, it should be confirmed by chromosome breakage studies using DEB or MMC. The family should be referred to a clinical geneticist and other sibs should be examined and offered DEB/MMC testing. Management thereafter depends on the age of presentation and the absence or presence of haematological abnormalities. All patients should have an ultrasound of the renal tract, hearing tests, and a haematological assessment that should include examination of the bone marrow. HLA typing in anticipation of possible bone marrow transplantation should be performed. An endocrinological assessment should also be made, especially if there is evidence of growth failure, and an ophthalmological assessment may show specific eye defects such as microphthalmia, almond shaped palpebral fissures, and epicanthic folds. Hand surgeons and plastic surgeons may consider correcting radial ray defects with a view to improving function and appearance.
Haematological management
If there is no haemopoietic defect at the time of diagnosis, haematological monitoring may only be required once per year but as the patient becomes older and develops haematological complications, haematologists play an increasingly central role. Many patients who develop bone marrow failure initially respond to supportive measures such as blood transfusions, androgens, and cytokines.54
Androgens, usually oral oxymethalone, are often used therapeutically because they enhance production and urinary excretion of erythropoietin and increase bone marrow cellularity.55 If the patient responds, the dose should be tapered over a period of three months to reduce side effects which include masculinisation, acne, hyperactivity, growth spurt followed by premature closure of the epiphyses resulting in short stature, deranged liver enzymes, hepatic adenomas, and potential risk of hepatic adenocarcinomas. Monitoring of liver function (liver enzymes, bilirubin, alpha-fetoprotein every two to three months and yearly liver ultrasound scans) is mandatory while on androgen therapy. Cytokines such as G-CSF and GM-CSF can improve haemopoiesis either in conjunction with androgens (particularly if the patient is neutropenic) or if androgens have failed. They should not be used in patients with clonal cytogenetic abnormalities and should be discontinued if such abnormalities develop while on therapy because of the potential risk of inducing leukaemia. Eventually most patients become refractory to therapy and the definitive treatment of choice is haemopoietic stem cell transplantation (SCT). Since it was found that cells from FA patients are more sensitive to radiation56 and conditioning agents such as cyclophosphamide,57 this has been carried out using greatly reduced doses, giving a two year survival rate of 70%.58 Alternative conditioning regimens using fludarabine and avoiding irradiation have shown promising results so far.59 For those patients without matched sibs, the prognosis for unrelated donors is less good at around 20–40% survival at two years and there is a higher risk of graft rejection in somatic mosaics.60
Recently, preimplantation genetic diagnosis (PGD) for parents of a previously affected child has been used to select embryos that were both unaffected with FA and HLA matched to their affected sib with a view to performing a SCT using umbilical cord blood. The procedure has been successful but required multiple attempts, led to wastage of otherwise healthy embryos (those that were unaffected but HLA mismatched), and is therefore still regarded as ethically controversial.61,62
Subsequent management
For those affected subjects who survive the haematological complications, follow up surveillance for solid malignancies becomes increasingly important.2 A study of secondary cancers in 700 aplastic anaemia patients who received an allogeneic bone marrow transplant highlighted FA as an independent risk factor for developing malignancy with a predicted risk of 42% at 20 years after transplant.63 Females should have annual gynaecological follow up to detect cervical and vulval tumours and both sexes should be kept under surveillance for oropharyngeal, oesophageal, and hepatic tumours. While there have been reports of increased radiation sensitivity in FA patients having radiotherapy for cancer treatment,64,65 this is not a consistent finding66 and cancer management based on the current limited evidence from FA patients should maximise surgical intervention while keeping chemotherapy and radiotherapy use to a minimum.
The multiple problems in early age, subsequent requirement for SCT, and continuing poor prognosis in survivors because of cancer susceptibility puts a great psychological strain on the FA patient and their family. Adequate psychosocial support and the involvement of a coordinated, multidisciplinary team with specialist knowledge are cornerstones to successful management of these patients.
Gene therapy
FA is an ideal candidate disease for gene therapy in view of the serious haematological complications requiring SCT and the likelihood that corrected FA cells have a selective advantage over defective ones, suggested by the high observed incidence of somatic mosaicism in FA patients. There have been successful attempts using retroviral mediated gene transfer in FANCC knockout mice which led to phenotypic correction of MMC sensitivity.67 Clinical trials so far in humans using retroviral vectors have been disappointing,68 but the recent use of lentiviral vectors in knockout mice has given promising results; efficient transduction could be attained using low viral dose, without cytokine prestimulation and minimal ex vivo manipulation, and may therefore be more successful than retroviruses in human therapy.69
GENETICS
Until recently, there were thought to be at least eight FA complementation groups determined by somatic cell hybridisation, FA-A, B, C, D1, D2, E, F, G. Six of the FA genes (FANCA, C, D2, E, F, G) have been cloned and it now appears that FANCD1 and possibly also FANCB are in fact BRCA213 (table 2). A further complementation group, FA-H, has subsequently been shown to belong to the FA-A complementation group.70FANCA mutations are the most prevalent, accounting for approximately two-thirds of all cases, FANCC and FANCG account for 25% of cases, FANCE and FANCF a further 8%, and the other groups less than 1%. None of the genes that have been cloned bear any homology to each other or to known DNA repair genes. Knockout mice have been generated for FANCA,71FANCC,72,73 and FANCG.74 All display a similar phenotype; they are viable, have no detectable developmental abnormalities, and do not develop any serious haematological complications or malignancies, although they do show a similar cellular phenotype to human FA patients, exhibiting spontaneous chromosomal instability and hypersensitivity to MMC. Both male and female mice show hypogonadism and impaired fertility.
The FA genes
THE FA GENES
FANCA was cloned in 19966,7 and it is one of the largest FA genes. Over 100 different mutations have been reported75–78 with 30% point mutations, 30% 1–5 base pair microdeletions or microinsertions, and 40% large deletions, removing up to 31 exons from the gene.77 Small duplications have also been reported.78 The large deletions often occur at specific breakpoints and have been shown to arise as a result of Alu mediated recombination.77,79–81 The heterogeneity of FANCA mutations and the fact that most patients are compound heterozygotes makes diagnostic screening for mutations difficult. Mutation analysis is less complex in populations where founder mutations have been described, such as the deletion exon 12–31 mutation, which accounts for 60% of mutations in Afrikaners.81
FANCA is ubiquitously expressed at low levels in all cells82 in both nucleus and cytoplasm,83 corresponding with its putative caretaker role within the cell.
FANCC was the first gene to be cloned by functional complementation in 19928 and compared to FANCA there is a much smaller spectrum of mutations. The most frequent is the intronic mutation IVS4+4A→T which leads to an altered splice site and deletion of exon 4; this is particularly prevalent in Ashkenazi Jews with a carrier frequency of 1 in 89 and is characterised by a severe phenotype with multiple congenital abnormalities and earlier onset of bone marrow failure.84,85 Interestingly, the same mutation is also the most prevalent FANCC mutation in the Japanese population (thought to be the result of a separate founder mutation), but the phenotype is less severe compared to Ashkenazis, suggesting that race specific modifying genes or environmental factors influence phenotype severity.86 Other common mutation sites are found in exon 1 and exon 14. FANCC is ubiquitously expressed and the protein has been localised to both cytoplasm and nucleus.87
FANCD1 has very recently been shown to be BRCA2 by sequencing the BRCA2 gene in cell lines from FANCD1 patients who were subsequently found to be compound heterozygotes.13BRCA2-/- cells exhibit many of the features of FA cells such as chromosome instability and hypersensitivity to cross linking agents,88,89 but null mice are not viable and no humans with biallelic germline BRCA2 mutations had been reported before, suggesting that this is not normally compatible with life in man. However, at least one of the mutations found in each of the two FANCD1 patients analysed is in the 3′ region of the gene, and it may be that those BRCA2-/- subjects expressing truncated BRCA2 protein with at least partial activity could be viable, and have a FA phenotype. Biallelic BRCA2 mutations have also been found in the FANCB reference cell line, suggesting the possibility of intra-allelic complementation or phenotypic reversion to wild type.70
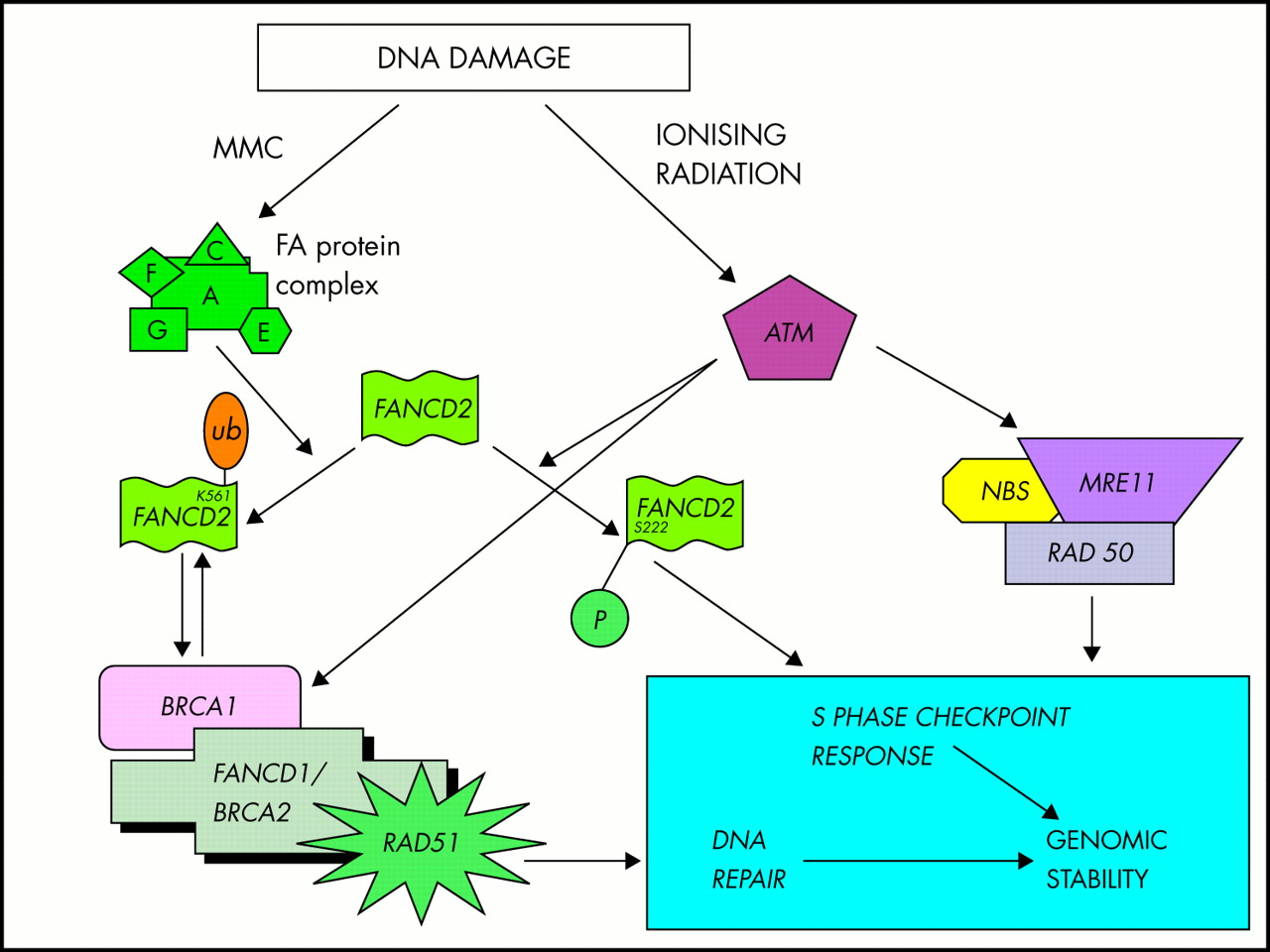
FANCD2 is the most recently cloned gene9 and although it accounts for only a very small number of FA families, it plays a key role in the FA pathway. Other FA gene sequences are conserved at the vertebrate level but FANCD2 is the only one conserved in lower organisms such as Drosophila melanogaster. The FANCD2 protein exists in two forms, FANCD2 short (S) and FANCD2 long (L), the long version being formed by the addition of a ubiquitin molecule to a lysine residue at position 561; upon monoubiquitination, FANCD2L is targeted to discrete nuclear foci where it colocalises with BRCA1.90 FA cells deficient for FANCD2 are sensitive to MMC but also have a degree of sensitivity to ionising radiation and have a defect in the ionising radiation inducible S phase checkpoint response similar to that seen in ataxia-telangiectasia (AT).15 When normal cells are exposed to ionising radiation, the AT protein product, ATM, phosphorylates FANCD2 on serine 222, activating this checkpoint response, a mechanism which is distinct and independent of MMC induced mono-ubiquitination.15 FANCD2 thereby provides a link between the FA protein complex and other pathways implicated in DNA repair by functioning at the intersection of two pathways, one involving MMC inactivation by the FA protein complex and the other involving ionising radiation inactivation by ATM.
The FANCE and FANCF genes were both cloned by functional complementation10,11 and between them account for less than 10% of all FA families. FANCG is identical to XRCC9,12 a human gene that corrects chromosomal instability and mutagen sensitivities in Chinese hamster ovary UV40 cells.91 All types of mutations apart from large deletions have been reported in FANCG.92
In a genotype-phenotype study of 245 FA patients from all complementation groups, a more severe cytopenia and higher incidence of leukaemia was detected in the FA-G group, there was an earlier onset of anaemia and a higher incidence of leukaemia in those patients with FANCA null mutations, and there was a reduced incidence of somatic abnormalities in the FA-C group excluding the IVS4+4A→T mutation.93 Thus, patients with FANCG mutations and FANCA null mutations appear to constitute a high risk group.
However, the broad similarities in phenotype between the different FA complementation groups make it likely that the FA protein products interact in a common pathway. This has been confirmed by yeast two hybrid and coimmunoprecipitation studies of FANCA, C, G, E, and F that have shown that each of these FA proteins interacts with at least one other. Initially FANCA and FANCC were shown to interact to form a complex and translocate into the nucleus.87 Subsequently FANCG, FANCF, and FANCE were all shown also to be part of this complex.94–96 It has also been shown that FANCC/A interactions are disrupted, not only in FANCC and FANCA cells, but also in FANCB, E, F, G cells and not in FANCD cells, suggesting that the FA protein complex acts upstream of FANCD2.97
Current evidence therefore suggests that a core complex composed of FANCA, C, E, F, G leads to activation of FANCD2S to FANCD2L by monoubiquitination (fig 3) and FANCD2L may then interact with BRCA1. FANCD2 also interacts independently with ATM activating the S phase checkpoint response. As the BRCA1 associated genome surveillance complex (BASC) includes DNA damage repair proteins such as ATM, NBS1, MRE11, BLM, and MSH2 (these genes are mutated in ataxia-telangiectasia, Nijmegen breakage syndrome, AT-like disorder, Bloom’s syndrome, and HNPCC respectively), FA may be linked at a molecular level to these other DNA damage repair disorders.98–100 The intriguing discovery that FANCD1 and possibly also FANCB are in fact BRCA2 indicates that the pathways involved in breast cancer susceptibility and FA are interconnected at more than one level.13,16
{kind=link}
{kind=link}
{kind=link}
A schematic diagram showing FA protein complex and the central role of FANCD2 in the DNA damage response pathway based on Taniguchi et al15 and Witt and Ashworth.16 DNA damage caused by cross linking agents such as MMC activates the FA protein complex leading to monoubiquitination of FANCD2, which interacts with BRCA1 to effect DNA repair. Damage caused by ionising radiation activates ATM which phosphorylates FANCD2 in addition to several other proteins to coordinate the S phase checkpoint response.
THE FA FUNCTIONAL PATHWAY
DNA repair in the FA cell
The exact mechanisms that lead to disruption of DNA repair in FA cells remain disputed.98,101 Repair of interstrand cross links (ICLs), induced by agents such as DEB and MMC, necessitates incision of DNA and both strands of DNA are damaged simultaneously so there is no available undamaged template. Double stranded breaks (DSBs) can be repaired by non-homologous end joining (NHEJ) where two DNA ends are ligated together at regions of little or no homology. Studies where plasmid extracts have been introduced into FANCC mutated cells have shown a reduced fidelity of non-homologous repair of blunt ended DSBs compared to normal cells, leading to a deletion prone phenotype which can be corrected by complementing the cells with wild type FANCC.102 Similar DSB repair error prone phenotypes have been described in FANCD1 defective cells,103 but also in other chromosome instability syndromes such as Bloom’s syndrome, ataxia-telangiectasia, and Werner’s syndrome.104 DSBs are generated during V(D)J recombination in normal cells and fidelity (but not frequency) of V(D)J recombination is reduced in FA cells.105 By contrast, homologous recombination by sister chromatid exchange does not seem to be defective in FA.106
In addition to abnormalities in NHEJ, it is possible that the detection of DNA damage in the FA cell is also impaired; there is evidence that the S phase cell cycle checkpoints are altered in that ICLs did not slow down or arrest FA cells in the S phase, with arrest instead occurring at the G2 checkpoint after completion of replication.107,108
These studies, taken together with emerging molecular evidence regarding the FA pathway, BRCA2 and FANCD2-BRCA1 interactions, and the well characterised chromosomal instability cellular phenotype, clearly indicate that FA proteins play an important role in DNA repair, which could either be direct involvement in repair of the DNA itself and/or in DNA damage signalling pathways mediating cell cycle checkpoints.
Other functions of FA proteins
In addition to defective DNA repair, a number of other possible causes of the abnormalities found in FA patients have been put forward, discussed in brief below.
Cell cycle control
The G2/M transition in the cell cycle is prolonged in FA cells38 and this is further increased if cells are exposed to cross linking agents or high oxygen concentrations.109 The G2/M checkpoint is associated with surveillance of the genome and repair of damage before progression to mitosis. This observation could be explained by the fact that FA cells fail to repair DNA damage efficiently and therefore arrest at G2.
Oxygen sensitivity
FA cells exhibit increased oxygen sensitivity and chromosome breakage is reduced in the presence of low ambient oxygen concentrations.110,111 Many DNA cross linking agents produce reactive oxygen species (ROS) and it is possible that the sensitivity of FA cells to cross linkers is the result of an impaired ability to counteract ROS. Clarke et al112 studied apoptosis in FA-C cells and showed that they behave like normal cells when exposed to MMC at 5% oxygen concentration and were hypersensitive to MMC at 20% oxygen, implying that it is the ROS generated by MMC, rather than the DNA cross link formation that causes toxicity in FA cells. Other lines of evidence come from yeast two hybrid studies which have shown that the FANCC protein interacts with NADPH cytochrome p450 reductase113 and FANCG with cytochrome p450 2E1 (CYP2E1),114 both enzymes that produce ROS.
Apoptosis and telomere maintenance
Various studies have shown apoptosis to be abnormally regulated in FA cells although the exact nature of the abnormality remains disputed.115 In one study, treatment of four FA lymphoblastoid cell lines with MMC led to a higher level of apoptosis,116 but other studies have shown higher levels of spontaneous apoptosis, lower levels after gamma irradiation, and no difference after MMC.117–119 Increased apoptosis may be related to inability of FA cells to repair damage or to defective FA protein interactions with other proteins in the apoptosis pathways.120,121 Accelerated telomere shortening has been detected in FA,122,123 but is also found in other forms of aplastic anaemia124 and in MDS.125 This could be explained by a haemopoietic stem cell undergoing a higher than normal number of cell divisions to generate mature end cells, but a recent study has shown that, in addition to replicative shortening, there is also a higher rate of breakage at telomeric sequences in FA cells, suggesting a defect in telomere maintenance.126
Haemopoiesis
In vitro bone marrow culture assays have shown defective haemopoiesis in FA127 and FA cells show altered levels of certain growth factors, such as reduced IL-6,128 GM-CSF,129 IL-1β,130 and increased TNF-α.131,132 These may affect haemopoiesis by altering the bone marrow microenvironment, leading to deregulation of cellular homeostasis, differentiation, and response to stress.
Haematopoietic tissue is one of the tissues most sensitive to DNA damage caused by radiation or cytotoxic therapy but while other DNA repair disorders such as xeroderma pigmentosum or ataxia-telangiectasia have an increased risk of certain malignancies, they do not have the same striking incidence of bone marrow failure seen in FA. It therefore seems likely that the FA proteins play a specific critical role in the maintenance of haematopoietic stem cells and that a combination of different functions of the FA pathway are implicated; the initial decline in haemopoietic stem cells leading to bone marrow failure could be the result of defective telomere maintenance and high rates of apoptosis. Then selective pressures may promote mutant clones developing as a result of the altered cytokine signalling environment against a background of genome instability leading to MDS and ultimately AML.133
The elucidation of the precise nature of these additional defects in the context of our current understanding of the interactions of the FA proteins remains a significant challenge, but it is clear that as well as interacting in a complex, at least some of the individual FA proteins interact with proteins in other pathways and are responsible for the cellular phenotypes described above (table 3).
Known FA protein interactions
FUTURE DIRECTIONS
With the characterisation of the main FA genes now accomplished, emphasis has moved to studying the overall function of the FA protein complex and interactions with other DNA repair proteins. Further protein interactions are likely to be found with the more recently cloned FA genes and a core DNA repair pathway involving the FA proteins has emerged. Many questions remain regarding functions of FA proteins outside DNA repair and how these affect phenotype. Correlation between the molecular defect, cellular defect, and clinical manifestation is an important task which will lead to better diagnosis and management of affected subjects with the ultimate goal of developing effective gene therapy. Improved haematological management in the form of specialised regimens for SCT in FA patients and the introduction of drugs such as fludarabine are leading to improved survival of FA patients, but this has resulted in larger numbers of homozygotes reaching the age where they are likely to develop solid tumours and further research will need to be done to determine optimum treatment for these malignancies.
SUMMARY
Fanconi anaemia is a heterogeneous condition that can present with a variety of congenital defects but invariably results in defective haemopoiesis which is the major cause of morbidity and mortality. There have been significant advances in the treatment of the haematological problems through the use of stem cell transplants, but there is a very high risk of oesophageal, vulval, and oropharyngeal tumours in survivors of the haematological complications. Seven main genes causing FA have been identified which will lead to the development of efficient and complete mutation screening, something that will become more important with the advent of preimplantation genetic diagnosis and the possibility of gene therapy. FA can be accurately diagnosed using DEB/MMC induced chromosome breakage which is highly specific. Over recent years much progress has been made in identifying the remaining FA genes, their interactions, and common pathways. The cloning of FANCD2, the identification of FANCD1 (and possibly also FANCB) as BRCA2, and development of a model for interaction of the other FA genes have linked the complementation groups together. Mounting evidence from cell studies together with the discovered link between FANCD2, BRCA1, and ATM are helping to elucidate the exact nature of the DNA repair deficit in FA and connect FA to the other DNA repair disorders.
Acknowledgments
We thank Professor C Mathew for his helpful suggestions and comments during the preparation of this manuscript and Professor E Gordon-Smith for supplying clinical photographs. MT is funded by Cancer Research UK.
Online resources for FA patients and their clinicians: www.fanconi.org Homepage of the Fanconi Anaemia Research Fund, the US patient support group. www.fanconi-anaemia.co.uk Homepage of the UK Fanconi Anaemia support group.