Article Text

Abstract
The pathogenesis of all forms of psoriasis remains obscure. Segregation analysis and twin studies together with ethnic differences in disease frequency all point to an underlying genetic susceptibility to psoriasis, which is both complex and likely to reflect the action of a number of genes. We performed a genome wide analysis using a total of 271 polymorphic autosomal markers on 284 sib relative pairs identified within 158 independent families. We detected evidence for linkage at 6p21 (PSORS1) with a non-parametric linkage score (NPL)=4.7, p=2 × 10-6 and at chromosome 1p (NPL=3.6, p=1.9 × 10-4) in all families studied. Significant excess (p=0.004) paternal allele sharing was detected for markers spanning the PSORS1locus. A further three regions reached NPL scores of 2 or greater, including a region at chromosome 7 (NPL 2.1), for which linkage for a number of autoimmune disorders has been reported. Partitioning of the data set according to allele sharing at 6p21 (PSORS1) favoured linkage to chromosomes 2p (NPL 2.09) and 14q (NPL 2.0), both regions implicated in previous independent genome scans, and suggests evidence for epistasis betweenPSORS1 and genes at other genomic locations. This study has provided linkage evidence in favour of a novel susceptibility locus for psoriasis and provides evidence of the complex mechanisms underlying the genetic predisposition to this common skin disease.
- psoriasis
- PSORS1
- epistasis
Statistics from Altmetric.com
Psoriasis is a chronic disfiguring skin disorder characterised by the triad of keratinocyte hyperproliferation, inflammatory infiltration, and vascular dilatation and proliferation.1 Chronic plaque psoriasis (vulgaris) is the most common (>90%), while other clinical forms include guttate, erythrodermic, and palmoplantar psoriasis.2 In 5-10% of psoriatics, a frequently debilitating arthritis may coexist.3 The estimated prevalence of psoriasis in the USA and northern Europe is 2%, yet is 10-fold lower among Japanese, west Africans, and mongaloids.2 The peak incidence occurs between the ages of 20 and 40 years, affecting males and females equally. While the aetiology of psoriasis remains unknown, current evidence indicates a central role for T lymphocytes in disease pathogenesis.1 Familial clustering,4 ,5together with the higher rates of concordance in monozygotic compared to dizygotic twins,6 suggests an important role for genetic susceptibility in the development of disease.
Predisposition to psoriasis is considered to be a complex genetic trait, with inheritance not following any simple Mendelian model.7 To date, seven independent genome wide scans have been performed to identify susceptibility genes for psoriasis, using both parametric and non-parametric approaches. It is now clear that a major locus for psoriasis susceptibility is located at chromosome 6p21.3 (PSORS1) with lod scores in excess of 3 being replicated by at least five reported studies, including our own.8-12 Significant evidence for linkage (lod score >3) has been identified for loci on chromosomes 17q (PSORS2),13 4q (PSORS3),14 1q (PSORS4),15 and 3q (PSORS5).16 Loci suggestive of linkage (lod score >1, <3) include those on chromosomes 2q, 8q, 16q, 19p, and 20p,8 ,9 ,13-17 but independent replication of these studies has proved difficult.
Because of the likelihood that multiple genes, acting either in an additive or multiplicative manner, may contribute to disease susceptibility, we have now undertaken a large sib pair family approach to map additional loci through a genome wide analysis. We report evidence for a novel locus at chromosome 1p, significant parental effect for genetic susceptibility at 6p21, and epistasis between a locus within the MHC and loci on chromosomes 2 and 14.
Materials and methods
FAMILIES
White families containing at least one informative affected relative pair were recruited primarily through two separate academic dermatology clinics in Glasgow and London, as well as additional dermatology centres. A standardised proforma, common to all sites, was used recording all essential clinical and family data including medical history, age of onset, and examination. Affected status was accepted only on the basis of active disease at the time of examination by a trained experienced clinician at each study site. Standard diagnostic criteria were used.2 Where affected status could not be assigned, subjects were retained in the genetic study as unknown phenotype. All subjects provided informed consent for this molecular genetic study, which was approved by local ethics committees.
SAMPLES AND GENOTYPING
Genomic DNA was extracted from 10 ml of peripheral blood using standard conditions.8 Genotyping was performed using a modification of the marker set described by Reed et al 18 and consisted of 271 markers and average heterozygosity of 79%. PCR was performed on MJ Research DNA Engines (10 μl reactions; 40 ng DNA, 1.5-2.5 mmol/l MgCl2, 0.2 mmol/l dNTPs (Pharmacia), 0.2 μmol/l primer, 0.2 units ofTaq polymerase (Advanced Biotechnologies) in a 1 × KCl PCR buffer).
Products were pooled and electrophoresed through 6% polyacrylamide gels (FMC) on an ABI 377 or 373 DNA sequencer. Semi-automated fragment sizing was performed using GENESCAN 2.1 software (PE-ABI) followed by marker peak detection using GENOTYPER 2.0 software (PE-ABI). Each genotype was reviewed manually to confirm the accuracy of allele designation. The Genetic Analysis System 2.0 (Alan Young, Oxford University, 1993-1995) was used to code allele sizes into whole numbers, to confirm Mendelian inheritance and to generate appropriate files for downstream analysis.
DATA ANALYSIS
Non-parametric analysis was assessed using the sib pair program of ANALYZE by J Terwilliger,8 ,19 while non-parametric multipoint analysis was performed with Genehunter 2.020 using the “all” statistic. Marker map positions were obtained from sex averaged maps available at the Genetics Location Database (http://cedar.genetics.soton.ac.uk/pub/) and the Marshfield clinic (http://www.marshmed.org/genetics/).
For analysis of epistasis and parental effect, the dataset was partitioned according to sharing of particular loci within families. For this the output for each family from Genehunter 2.0 was used.21 Those families with aNPL score >0.8 for a particular region were designated NPLPOS (66 families, 136 ASPs) and those families with aNPL score <−0.8 were designated NPLNEG (22 families, 22 ASPs). Remaining families were not included in the further analyses. This method of partitioning was performed for loci located on 6p21 and 1p. NPLPOS families for chromosome 6p21 and chromosome 1p were assessed using the SIBDES program of the Genetic Analysis System 2.0 which also includes a 1:0 distribution for each parental sex, allowing the comparison of sharing for alleles derived for each parent.
Results
We performed a genome wide screen using 284 ASPs (222 independent sibships22) identified within 158 nuclear families (table 1). Of the 472 affected subjects (258 female, 214 male) the mean age of onset was 15.8 (2-70) for females and 19.2 (1-60) for males. We used a panel of 271 markers distributed across the 22 autosomes with an average intermarker distance of 13 cM. Fig 1shows the non-parametric linkage (NPL) score obtained using the Genehunter 2.0 program, plotted for each chromosome, for all families and also for chromosome 6p21 NPL positive (NPLPOS) and NPL negative (NPLNEG) families.
Details of study cohort
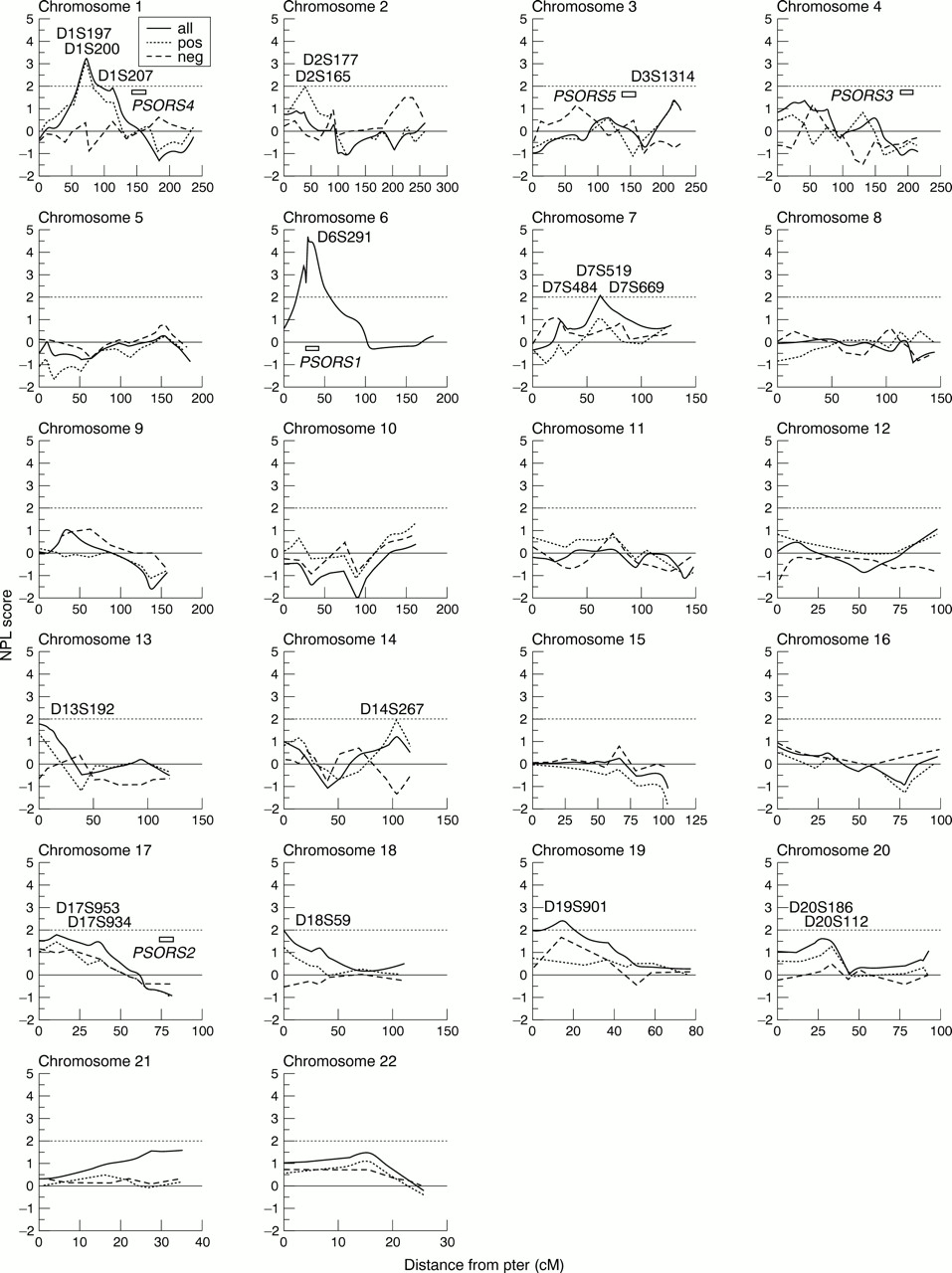
Non-parametric multipoint linkage analysis in 284 psoriasis affected sib pairs performed using Genehunter. This includes all families (solid line), families which obtain positive NPL scores for the MHC region of chromosome 6 (dotted line), and families which obtain a negative NPL score (dashed line). Markers are shown for regions of interest and previously identified regions submitted to OMIM are marked. Map positions obtained from the sex averaged maps compiled by J Weber (http://www.marshmed.org/genetics). A full marker list is available from the corresponding author.
NOVEL LOCUS FOR PSORIASIS SUSCEPTIBILITY AT 1P
Highly suggestive evidence for linkage was observed on chromosome 1p, with a peak between markers D1S197 and D1S200 (NPL=3.6, p=1.9 × 10-4). This observation approximates to the 5% genome wide significance level for a 10 cM scan.23
A total of 16 regions generated nominal evidence for linkage (NPL >1.0), and these were distributed across 14 chromosomes. Of these, five regions reached or exceeded NPL scores of 2, including chromosome 7 (NPL=2.1, p=0.0167), a region showing clustering of autoimmune diseases,24 between D7S519 and D7S669, chromosome 18 (NPL=1.97, p=0.025), between D18S59 and D18S1150, and chromosome 19 (NP=2.4, p=0.008), between D19S901 and D19S221.
These regions and others previously reported as putative susceptibility loci for psoriasis were also investigated using a two point test for allele sharing within the ANALYZE package (table 2). Probability values=0.05 were observed at D3S1314 (p=0.05), D6S300 (p=0.03), D7S519 and D7S669 (p<0.03), D18S59 and D18S1150 (p=0.04).
Summary of potential psoriasis susceptibility loci : non MHC loci
EVIDENCE FOR PARENTAL EFFECT AT 6p21
Genome wide, the most significant evidence for linkage was observed at chromosome 6p21, spanning the region containing the human MHC. Genehunter generated the strongest evidence for linkage for the markers D6S273 and D6S291 (NPL=4.7, p=2 × 10-6) (fig1). Comparable results were observed using a two point sib pair analysis (p=3.4 × 10-8) (table3).
Markers within or flanking the MHC on 6p21 (PSORS1)
We next examined those families designated NPLPOS for the region at 6p21, for sharing of alleles IBD between affected sib pairs according to parental origin. A highly significant difference for allele sharing was detected for the markers D6S276, tn62, D6S273, D6S291, and D6S300, with up to 92% of allele sharing between ASPs being paternally derived (p value of 0.004 for alleles at D6S273 in a test for heterogeneity using a 2 × 2 contingency table chi square test) (table 4). For comparison we also analysed those families found to be NPLPOS for the next most significant region for linkage, namely that between markers D1S197 and D1S200 at chromosome 1p. No significant parental effect was observed for any of the markers from chromosome 1p (fig 2).
Sharing between affected sib for paternally and maternally derived alleles on chromosome 6p

{kind=link}
{kind=link}
P values plotted from GAS 2.0 for markers from the two linkage regions at chromosomes 6p21 and 1p. Black boxes represent p values for sharing of the paternally derived alleles and white boxes the maternally derived alleles. (Left) Parental origin for NPLPOS families for the 6p21 linkage region (PSORS1). (Right) Parental origin for NPLPOS families for 1p linkage region.
EVIDENCE OF EPISTASIS BETWEENPSORS1 AND OTHER LOCI
In the light of evidence for multiple loci contributing to psoriasis susceptibility within our cohort, we sought to reduce the heterogeneity of the data set by partitioning based on sharing of alleles at 6p21, the region showing the greatest evidence for linkage. Families providing either positive or negative NPL scores were reanalysed using Genehunter. NPLPOS families provided NPL scores above 2 for regions on chromosome 2p (NPL=2.09, p=0.019) and chromosome 14q (NPL=2.0, p=0.026), both within regions previously reported in genome wide scans.8 ,17 In contrast, NPLNEG families made no contribution to the 1p and 19p linkage scores.
Discussion
The pathogenesis of psoriasis is complex, with both genetic and environmental factors contributing to the development of the disease. This and previous genome scans have identified a major psoriasis susceptibility locus within the MHC (PSORS1) on chromosome 6p21, which is estimated to contribute up to 50% of the familial clustering underlying psoriasis.8-12 Hence, non-MHC loci must exist but individually these are likely to have a small genetic effect, the detection and replication of which is likely to require both the use of large datasets and more dense genetic marker panels. In this study, we have used a much larger number of affected relative pairs, with an optimised polymorphic STS marker set. Our genome scan provides both confirmation of the MHC susceptibility locus (PSORS1) and evidence for a locus on the short arm of chromosome 1.
Three consecutive markers, D1S197, D1S200, and D1S207 at chromosome 1p contributed to a significant NPL score of 3.6, which reaches published criteria for genome wide significance in the search for loci contributing to complex disease susceptibility.23Of interest, in our initial genome scan, using an independent cohort of psoriasis kindreds, marker D1S197 generated a single point sib pair analysis value p=0.018.8 Neither study detected evidence for linkage disequilibrium using the transmission disequilibrium test for the markers described. This may reflect a spacing of markers too broad to detect LD with a disease gene or the absence of any such association. A number of immune related genes have been mapped to 1p. Intriguingly, the region includes the gene encoding EPS15, a highly specific intracellular substrate for the EGF receptor which itself is known to be overexpressed in psoriatic epidermis.25 The presence of positional biological candidate genes combined with the replication of findings from a previous genome analysis suggest this region is now worthy of more detailed investigation.
We also provide further confirmation for the major role of a gene or genes located within the MHC at chromosome 6p21 to psoriasis susceptibility. Using an ancestral haplotype strategy, the minimum interval for the PSORS1 locus has been mapped to a 300 kb genomic fragment between the class 1HLA C gene and the Sgene,26 ,27 which encodes the late epidermal differentiation protein, corneodesmosin.28-30 The genomic sequence of this region has recently been published and a number of novel genes identified, each of which represents a positional candidate for PSORS1.31 This region is currently the focus of intense investigation using a combination of genetic and functional studies. Of the other regions of putative interest identified in this study, intriguingly markers on chromosome 19p13.3 (D19S901) lie within a region paralogous to the MHC, a region assumed to have arisen as an early duplication event.32 Identification of thePSORS1 gene variants will enable further assessment of this finding and in particular a search for sequence homology between the two regions.
The pivotal role of PSORS1 justifies more detailed exploration of its potential interactions with other susceptibility loci in the aetiology of psoriasis. We partitioned families displaying evidence of linkage for markers (table 4) on chromosome 6p21 as this region has been replicated in several independent genome wide scans and has been refined to a limited genomic region. This approach has the potential to separate heterogeneous contributions to psoriasis. For two independent chromosomal regions, 2p and 14q, suggestive evidence for linkage (NPL score ⩾2.0) was detected in the NPLPOS group, indicating a potential epistatic effect. However, these data must be interpreted cautiously, both because of the multiple statistical tests applied (stratification) and the relatively small sample size from which the results are derived.1However, it is of interest that both of these regions have previously been implicated as putative susceptibility loci.8 ,17Further study of larger cohorts (in excess of 1000 sib pairs) will be required to enable detailed genetic analysis of these regions with a smaller contribution to the familial clustering in psoriasis.
We next examined the contribution of PSORS1according to parental sex. Previous segregation analysis data showed a significantly higher penetrance of psoriasis if the father was either affected or a presumed gene carrier.7 Distortion of sharing of paternally derived alleles at markers on chromosome 6p has been suggested previously by Burden et al 33 and is now shown in a larger data set. Possible mechanisms that may account for these observations include genetic imprinting of a susceptibility allele, and similar findings have been proposed for type 1 diabetes mellitus susceptibility locus IDDM2.34 ,35 Whatever the mechanism, the strength of this finding would suggest that parental effects should be considered in regard of the fine mapping and characterisation of the genetic variant(s) contributing toPSORS1.
Despite the use of a larger data set, which readily detected linkage toPSORS1, we did not find support for linkage for a number of previously identified psoriasis susceptibility loci (table 2). Replication of findings from independent genome wide scans has rarely been achieved to date. It has been suggested that confirmation of linkage will require yet larger study cohorts than those used in the initial scan. Furthermore, the marker set used in this study was not developed to enable optimal replication of previously reported loci. Finally, the possibility of both false positive and negative findings, particularly in association with multiple statistical testing, has been widely discussed.36
We can conclude that the molecular genetic basis of susceptibility to psoriasis is complex, with heterogeneity, multiple genes, and possible epistatic interactions all contributing. The susceptibility locusPSORS1 appears to represent the major locus in this disorder and fine scale mapping of this region is now feasible. This study points to the utility of the reduction of heterogeneity in pooled data sets that will be achieved through future analysis, based upon the presence or absence of the genomic variants that contribute toPSORS1. The elucidation of the molecular mechanisms underlying such genetic interactions must await the identification and functional analysis of the relevant genes.
Acknowledgments
We are most grateful to all the families with psoriasis who have so willingly participated and encouraged us with these studies. We thank the many UK dermatologists who have provided details of patients under their care. We acknowledge statistical advice from J Terwilliger and excellent technical support from Roshna Mistry. This work was supported by grants from The Wellcome Trust (RCT, DT, and JNWNB), the Psoriasis Association of Great Britain (RCT), and the Dunhill Medical Trust (JNWNB). NB is a Medical Research Council Training Fellow.