Article Text

Abstract
Spinal muscular atrophy (SMA), a clinically and genetically heterogeneous group of neuromuscular diseases, is a disorder of motor neurones characterised by degeneration of spinal cord anterior horn cells and muscular atrophy.
SMA is an autosomal recessive disorder with a carrier frequency of about 1/50. Three candidate genes, the survival motor neurone (SMN) gene, the neuronal inhibitory protein (NAIP) gene, and the p44 (subunit of basal transcription factor TFIIH) gene, have been considered as genes involved in this condition. The region spanning these genes has a complex organisation including duplications, repetitive sequences, truncated genes, and pseudogenes, which makes molecular analysis of this condition difficult. Although deletions have been found in the majority of SMA patients, a few microrearrangements (like duplications, missense mutations, microdeletions, and gene conversions) localised in the telomeric form of the SMN gene have also been reported.
The function of the protein encoded by the SMN gene is still not fully understood but recent studies have indicated that it is found intracellularly in gems, novel nuclear structures. Its interaction with other proteins suggests a role in mRNA processing and metabolism. Whether the NAIP gene protein and other apoptosis associated proteins are directly involved in the initial stages of neurone degeneration and apoptosis, or acting downstream on the pathological pathway, has been difficult to determine. Further studies will be required to elucidate possible functional interactions between these proteins.
- SMA
- SMN
- NAIP
Statistics from Altmetric.com
The term spinal muscular atrophy (SMA) comprises a clinically and genetically heterogeneous group of diseases characterised by degeneration and loss of the anterior proximal horn cells in the spinal cord, and sometimes also in the brainstem nuclei, resulting in muscle weakness and atrophy.1 Most cases of SMA are inherited as an autosomal recessive trait, although dominant families are known. This neuromuscular disease is the second most common lethal autosomal recessive disorder in white populations with an overall incidence of 1 in 10 000 live births and a carrier frequency of about 1/50.2
In 1891, Guido Werdnig (1844-1919), an Austrian neurologist from the University of Graz, described two brothers with proximal SMA.3 Two years later, Johan Hoffmann (1857-1919), a German neurologist from the University of Heidelberg, described four cases belonging to two families where some of these children did not show the fatal course of the disease.4 In the mid fifties, Kugelberg and Welander5 6 described 12 patients belonging to eight sibships with an onset of the disease between 2 and 17 years of age. All were able to walk at least eight to nine years after the symptoms started.
This resulted in the classification of childhood onset SMA into three types, distinguished on the basis of clinical severity and age of onset. Type I (Werdnig-Hoffman disease) is the most severe form with onset within the first six months of life. Children are never able to sit without support and usually die within two years. Type II (Dubowitz disease) is the intermediate form of SMA and has an onset before the age of 18 months. Children are unable to stand or walk and death usually occurs after the age of 2 years. Type III (Wohlfart-Kugelberg-Welander disease) is a mild, chronic form with onset after the age of 18 months.
A new era of understanding the molecular basis of SMA began in 1990. Linkage studies with microsatellite markers using consanguineous families7 and large numbers of small families segregating different forms of SMA8-10 showed that the mutation in all three forms of SMA mapped to the same region of chromosome 5q.
Radiation hybrid mapping11 and construction of a non-chimeric YAC contig12 13 reduced the candidate region to approximately 400 kb. As the region of interest narrowed, the complexity, that is, repetitive sequences, pseudogenes, retrotransposable elements, deletions, and inverted duplications,14-16 became clearly evident and explained the difficulties encountered in attempting to generate physical maps of the region.
In the January 1995 issue of Cell, two different SMA candidate genes (survival motor neurone gene (SMN)17 and neuronal apoptosis inhibitory protein gene (NAIP)18 were described and, interestingly, each was present in at least two copies. More recently, a third gene, encoding p44 (a subunit of the basal transcription factor IIH (BTF2p44)), has also been found to be associated with SMA type I.19 20
Characterisation of the genes
Both copies of the SMN gene contain nine exons. The two copies are named telomeric (SMNT) and centromeric (SMNC) or CBCD541. They differ only in eight nucleotides (five are intronic and three are exonic, located within exons 6, 7, and 8),17 21 22 thus enabling them to be distinguished either by SSCP (single stranded conformation polymorphism) analysis17 or by enzyme digestion of PCR products.23
Although both copies are ubiquitously expressed mainly as a 1.7 kb transcript,17 alternative splicing generates minor isoforms of the SMN protein.17 24 The majority of transcripts (∼90%) from the SMNT gene are full length and about 10% of transcripts lack exon 5. The SMNC gene produces a variety of different isoforms, including transcripts without exon 5 or 7, transcripts lacking both exons, as well as full length SMN transcripts. The full length transcript accounts, surprisingly, for 20-30% of the total transcripts from the SMNCgene.17 24
The neuronal apoptosis inhibitory protein gene (NAIP) was so named because of high homology between exon 5 and 6 and a portion of a baculoviral gene product, inhibitor of apoptosis protein (IAP). This homology raised the intriguing possibility that SMA was a disorder of unrestrained apoptosis. Later, however, gene homology studies and expression studies showed many differences in gene structure and expression patterns and concluded that NAIP and IAP both restrain apoptosis, but in different fashions.25 The NAIP gene contains 17 exons.22 A telomeric functional copy (NAIPT), Ψ (pseudo) NAIP copy, and several truncated copies are present in the SMA region.18 Exons 5 and 6 are exclusively present only in the NAIPT copy. A PCR based assay for these exons enables one to distinguish the presence or absence of an intact NAIPT copy.18
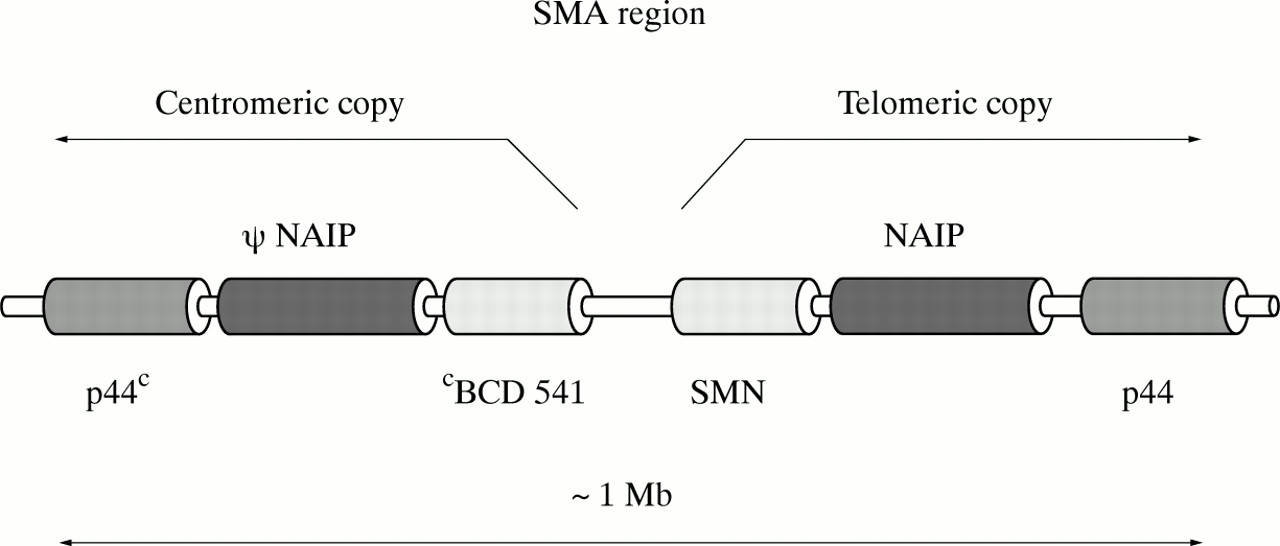
A single copy of the gene encoding p44, a subunit of the basal transcription factor TFIIH, was characterised and localised in 1994-1995.26 27 Very recently, new data about the p44 gene in relation to its potential involvement in SMA were described by Carter et al 19 and Burglenet al.20 They showed that the p44 gene is present in the SMA region in two copies, as a telomeric and centromeric form, and both are transcribed. The full length gene contains 16 exons. Four nucleotide substitutions enable the telomeric p44T and the centromeric p44C genes to be distinguished. Burglen et al 20showed that p44 is also subject to alternative splicing. The probable alignment of genes in the SMA critical region is depicted in fig1.
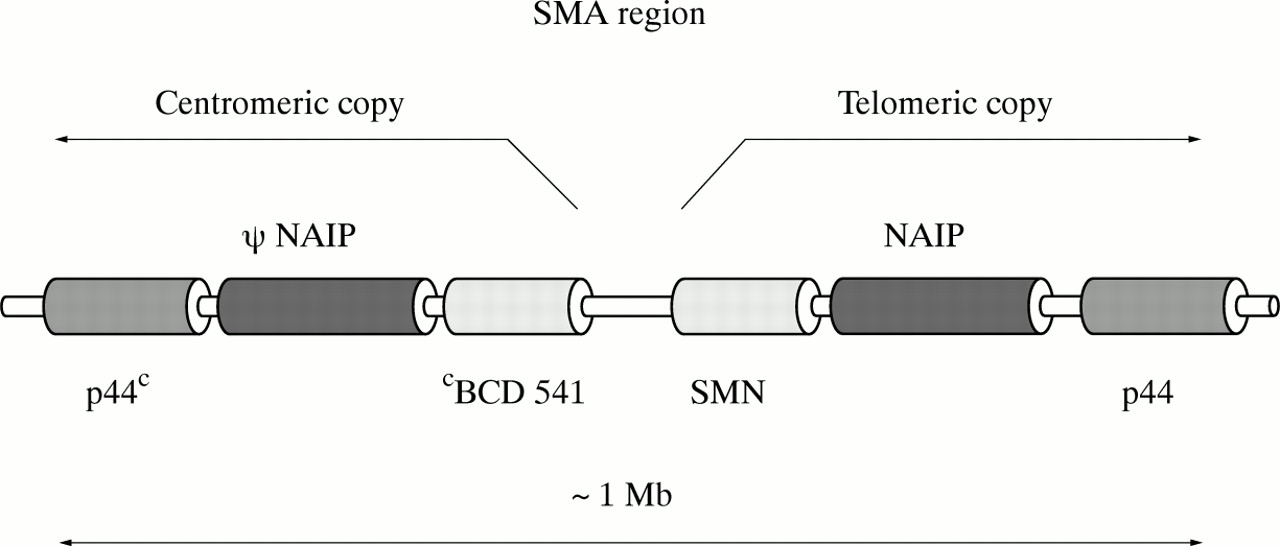
The probable alignment of genes in the SMA critical region (based on Burglen et al20).
Which gene(s) causes the disease?
It is important to determine whether one of these genes in particular plays a more significant role in the development of spinal muscular atrophy.
It was clearly documented by Lefebvre et al,17 Velasco et al,28 Rodrigues et al,29 30 and Cobben et al 31 that up to 95% of patients suffering from different forms of SMA are homozygously deleted for both telomeric copies of the SMN gene. In the detailed initial study by Lefebvreet al,17 the patients who did not have homozygous deletions were shown to have point mutations in the SMN gene. Further studies32-35 also confirmed the involvement of missense mutations in the SMN gene as part of the mutation spectrum. Interestingly, Talbot et al 33 illustrated a clustering of all known missense mutations within a small interval from codon 262 to codon 279 in the SMN gene. This region has been shown to be 100% conserved between humans, mouse, and rat33 and therefore can be considered to be an important functional domain of the SMNT protein. In addition, further small SMN gene rearrangements like microdeletions17 36-38 and duplications39have also been detected in SMA patients. The topographic distribution of various microrearrangements and missense mutations identified in the SMN gene are listed in table 1 and depicted in fig2.
Mutations involved in SMA
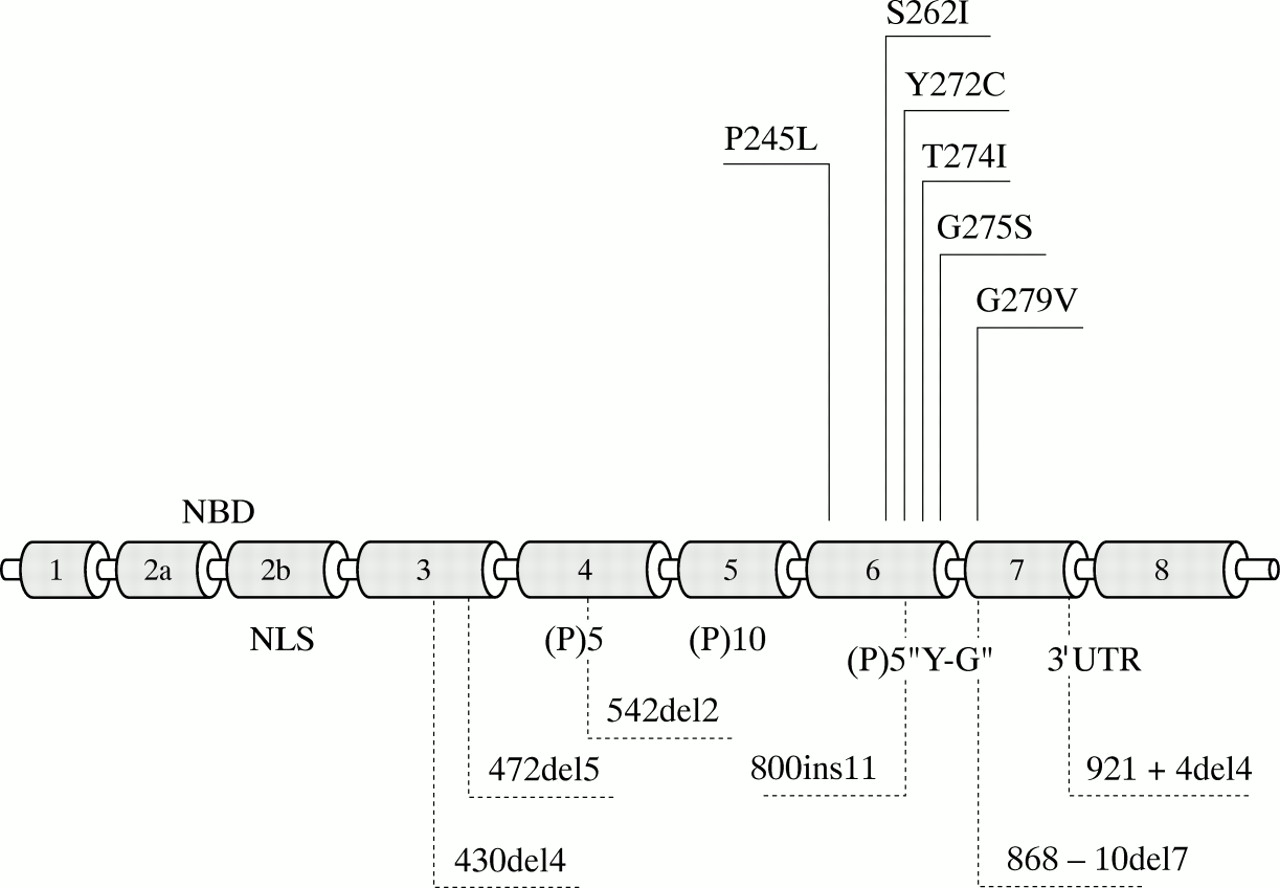
The topographic distribution of the SMN protein domains encoded by the SMN gene and various microrearrangements and missense mutations identified in the SMN gene. NBD=nucleic acid binding domain, NLS=nuclear localisation signal, (P)5=stretch of five prolines, (P)10=stretch of 10 prolines, “Y-G”=“Y-G box”. See text and table 1 for details and references.
In contrast, as documented in table 1, deletion of both copies of NAIPT was observed initially in 45% of SMA type I patients.18 Slightly higher proportions (62-69%) were reported from the studies conducted by Velasco et al,28 Rodrigues et al,29 and Hahnen et al.40 These groups and also the original description by Roy et al 18 showed that fewer SMA type II and III patients (12-18 %) were deleted for NAIP. Homozygous deletion of the NAIP gene is the only mutational event so far detected in this gene. It was well documented by Somerville et al 41 that when both the SMN and NAIP genes are homozygously deleted, there is a five-fold increased risk of type I SMA. Although in most cases if a NAIPT deletion is detected, the patient is also deleted for the SMNT gene, there are exceptions. A NAIPT deletion with retention of at least one copy of the SMNT gene was found in about 2% of SMA type I patients.28 30 These studies, however, did not analyse the sequence of the entire SMN gene to determine if point mutations or other small intragenic rearrangements were involved.
The role of the p44 gene in the clinical manifestation is still questionable. Both original papers19 20 showed that the telomeric version of the p44 gene is deleted in about 15% of all SMA types. A combined analysis of the entire SMA region, however, indicated that p44 gene deletions are part of a large scale deletion involving the SMN and NAIP genes as well. Carter et al 19 did report two type III SMA sibs deleted for both copies of the p44T gene while retaining at least one of their functional copies of the SMN and NAIP genes. Further studies are needed to determine if p44 is critical for the development of SMA.
From all these studies, one could conclude that deletions of this SMA determining region would invariably result in an SMA phenotype. However, Wang et al 42 clearly documented SMNT homozygous null alleles in parents of SMA patients. Even more surprising observations were published by Hahnenet al,40Burghes,43 and Cobben et al.31 Both groups identified a pair of haploidentical sibs, both homozygously deleted for SMNTwith phenotypical variation, one fully expressing the SMA phenotype and the other being asymptomatic. Similarly, Velasco et al 28 and Rodrigues et al 30 showed that about 1-2% of phenotypically normal SMA carriers are deleted for NAIPT. Carteret al 19 also found a homozygous deletion of the telomeric copy of the p44 gene in one normal subject.
Genotype versus phenotype
How, then, does the knowledge of the different genes involved explain the range of phenotypes observed in people affected with spinal muscular atrophy? Absence of exons 7 and 8 of SMNT detected by PCR was found in more than 95% of all types of SMA patients, but exact correlation between the extent of the apparent deletion and the clinical severity has not been established. What about the possible involvement of the centromeric counterpart of the SMN gene,CBCD541, in the pathophysiogenesis of SMA?
Because homozygous deletion of both copies of CBCD541 in the presence of at least one copy of the SMNT gene does not cause the SMA phenotype, it was originally thought that the centromeric copy was non-functional. CBCD541 was shown, however, to produce 20-30% of its transcripts as full length functional protein. Lefebvre et al 17 were the first to suggest a possible inverse relationship between the number ofCBCD541 copies and the severity of the spinal muscular atrophy. This suggestion was later confirmed by Campbellet al 44 who, using pulsed field gel electrophoresis, clearly showed a higher number ofCBCD541copies in type II and III SMA patients compared with type I patients. Valesco et al 28 had performed densitometric analysis in parents of type II and III SMA patients and showed that they carried three or more copies of CBCD541. The mechanism(s) for the presence of extra copies of CBCD541 has not been fully eludicidated but gene conversion or duplication have been hypothesised as likely possibilities.45 These groups also concluded that type I SMA is, in the majority of cases, characterised by physical deletion of the SMNT gene while the mutations in type II and III SMA are replacements of the telomeric copy by the centromeric form.
A more detailed examination of the SMNC copy number conducted by McAndrew et al 46indeed showed a shift in distribution with the milder phenotype having a higher number of SMNC copies. An unusual example was described by McAndrew et al 46where one asymptomatic subject who had homozygous deletion of both copies of the SMNT gene had four copies of SMNC. Interestingly, this group reanalysed the haploidentical sibs described by Burghes.43 These sibs varied clinically from expressing SMA to mildly affected/unaffected and both had three copies of SMNC and no copy of SMNT. Therefore, at a simplistic level, study of the copy numbers of SMNC and SMNT do not explain the phenotypic difference. Burghes,43 however, proposed that two different functional SMNC alleles exist: one such allele, created by gene conversion (but not including the 5′ end of SMNT), can modify the phenotype, whereas the other allele , SMNC in its original physical context or formed by extended conversion through the entire SMNT gene, cannot modify the phenotype.
Partial gene conversion events have been also shown to be the explanation for about 5% of SMA patients who have absence of exon 7 of SMNT on PCR but are positive for exon 8.43From the description of all these different types of alleles, Burghes43 has classed them into mild and severe. He proposed that type I SMA is caused when two severe SMA alleles are present, type II SMA when one mild allele is present, and type III SMA when two mild alleles are present. A modified version of his model is presented schematically in fig 3.

{kind=link}
{kind=link}
{kind=link}
Schematic overview of the possible SMN alleles causing severe or milder forms of SMA (modified from Burghes et al43). SMNC/T: allele retaining 5′ end of SMNC with exons 7 and 8 originating from SMNT. SMNT/C: allele retaining 5′ end of SMNT with exons 7 and 8 originating from SMNC. SMNT/C/T: rare conversion allele in which exon 7 of SMNT is converted to SMNC. SMNT#: SMNT allele with small rearrangements like missense mutation, microdeletion, or duplication.
Diagnostic testing
Relatively simple DNA tests based either on SSCP analysis17 or PCR followed by restriction enzyme digestion23 enables confirmation of a suspected clinical diagnosis of SMA or prediction of the outcome of a pregnancy in families with a history of SMA (as reviewed by Zerreset al 47 and Brahe and Bertini48). The absence of both copies of the SMNT gene is a very reliable and powerful diagnostic assay for the majority of SMA patients, confirming their clinical SMA phenotype. Zerres et al 47suggested that as all NAIP deleted patients are also deleted for SMNT, NAIP testing does not need to be part of routine diagnosis. However, there are about 2% of patients that are deleted for NAIP but not both copies of SMNT.28 30One could therefore test with NAIP if patients are not deleted for SMNT.
With respect to linkage analysis in prenatal diagnosis, opinions are varied. Many diagnostic laboratories still perform linkage analysis in addition to direct SMNT mutation analysis in families where the previously affected child lacks both copies of SMNT. The basis of this appears to stem from the fact that there are people in the normal population who lack both copies of SMNT but are not clinically affected. Therefore one of the carrier parents may possess two deleted alleles, but only one when passed to the next child will result in SMA. The only way to confirm that the same allele has been inherited from both parents, and therefore that the next pregnancy is at risk of SMA, is to include linkage analysis when the fetus lacks both copies of SMNT. Linkage analysis is the only option in the families where no deletion has been observed but the clinical findings are consistent with 5q SMA.
On the other hand, the PCR based assay for determining the presence or absence of SMNT is not quantitative and therefore it cannot detect subjects with heterozygous absence of the SMNT gene. Practically, this test therefore cannot identify SMA carriers, or distinguish between a non-5q SMA-like patient and a compound heterozygote 5q SMA patient with a point mutation in one allele and absence of the other. The development of a quantitative assay was therefore of high priority.
DOSAGE ANALYSIS
The first attempts to estimate the copy number used the densitometric assessment of the SMNT/SMNCratio28 49 or solid phase minisequencing.50The drawback of these methods was that they were not suitable for SMA carrier analysis or detection of compound heterozygotes because they relied on the ratio of SMNT/SMNC without the use of an external standard. Incorporation of two external controls became an essential part of a novel dosage analysis designed by McAndrew et al,46 which improved the diagnostic power to detect absence of one copy of the SMNT gene.
An example of the successful application of the technique to confirm the diagnosis of a possible case of severe SMA was given by Parsonset al.38 The patient was directly tested and found not to have the homozygous deletion of SMNT found in about 95% of patients. The quantitative PCR assay was performed and the patient was found to have lost one copy of SMNT. Subsequent sequencing resulted in the identification of a 2 bp deletion in exon 4. This illustrates the value of the methodology, not only for carrier detection but also for confirmation of diagnosis in patients who are compound heterozygotes with loss of one SMNT allele and a mutation in the second.
Another strategy for determining the copy number of the SMNC and SMNT genes was designed by Campbellet al.44 They used pulsed field gel electrophoresis and interpretation of the hybridisation pattern of SMN and NAIP probes for their analysis. Although an elegant technique, it is not likely to be routinely used in a DNA diagnostic laboratory.
Although carrier detection studies have focused on the analysis of the SMNT copy number, SMNC copy number is also determined in the procedure. The exact interpretation of the SMNC analysis with respect to carrier status prediction is not clear at present.
From genes to proteins
SMNT PROTEIN CHARACTERISATION
When the SMN gene was initially identified in 1995 by Lefebvreet al,17 it had no known homology to existing proteins, and the function and subcellular localisation were unknown. Lefebvre et al 17 did identify a stretch of five prolines in exons 4 and 6 and 10 prolines in exon 5 which might have an important role in the folding of the SMN protein, thus initiating the elucidation of several functional domains. DiDonato et al 51 showed a putative nuclear localisation signal (NLS) encoded in exon 2b. Finally, Talbot et al 33 described the clustering of missense mutations within exons 6 and 7 (the “Y-G box”), which is part of an evolutionarily highly conserved region, and is an important functional domain involved in mRNA metabolism. Further insight into functional characterisation of this domain was elucidated by Lorsonet al. 52 They identified a small, 30 amino acid region which directly mediates SMN self-oligomerisation. The authors also compared wild type to mutant SMN proteins of type I, II, and III SMA patients and found a direct correlation between oligomerisation and clinical type. Recently, Lorsonet al 53 identified a potent non-sequence specific nucleic acid binding domain (NBD) encoded by exon 2 of the SMN gene. Interestingly, they tested the previously reported missense mutations S262I34 and Y272C17 and found that RNA binding activity dropped two to three-fold. The topographic distribution of the above mentioned SMN protein domains are depicted in fig 2.
SUBCELLULAR LOCALISATION
The first information about the subcellular localisation of the SMN protein was described by Liu and Dreyfuss in 1996.54In searching for SMN interacting proteins using a yeast two hybrid screen, they found that the SMN protein interacts with the RGG box region of hnRNP U protein, with itself, with fibrillarin, the small nucleolar RNA binding protein, and with several novel proteins as well.
Using antibodies raised against the SMN protein, Liu and Dreyfuss54 localised it into discrete dot-like novel nuclear structures named gems (gemini of coiled bodies), as well as finding it in the cytoplasm. Gems are located in close proximity to coiled bodies which are conserved subnuclear structures. Both are disassembled and reassembled during the cell cycle in a similar fashion. Although the specific functions of coiled bodies are as yet unknown, their components suggest their possible role in the metabolism of snRNPs (small nuclear ribonucleoprotein particles) and in pre-mRNA processing. As documented by Coovert et al,55 the number of gems is significantly reduced in type I SMA patients (100-fold reduction compared to unaffected controls) and this also correlates with the clinical severity of SMA.
Further detailed studies from Liu et al 56 showed that the N-terminal region of the SMN gene product is tightly associated with a novel protein SIP1 (SMN interacting protein). The SMN-SIP1 complexes are large and contain several U snRNP proteins. One subclass of these proteins is known as the common proteins as they associate with all U snRNAs and contain a related Sm domain. Most of the common proteins bind recombinant SMN protein in vitro.56 Antibodies directed against the SMN-SIP1 complex strongly interfere with the cytoplasmic assembly of the common snRNP proteins, with spliceosomal snRNAs, and with the import of the snRNP complex into the nucleus.57 Based on these findings, Fischer et al 57therefore suggested a role for SMN and SIP1 in spliceosomal snRNP biogenesis and a functional defect in this assembly may, therefore, be the cause of SMA.
EXPRESSION STUDIES
SMN gene expression studies58 59 showed that both forms of the gene were expressed in various non-CNS tissues (including heart, liver, muscle, lung, thymus, pancreas, kidney, and also lymphoblastoid cell lines) as well as CNS derived tissues. Lefebvreet al 58 showed that the SMN protein was widely but unevenly present in all parts of the brain and even within different motor neurone cells. An increased level of the protein found in the spinal cord inversely correlated with the severity of the disorder.
Lastly, it should be asked why such a devastating effect is observed only in the motor neurone cell, when the SMN protein is expressed in various non-CNS derived tissues. One possibility which was named “the threshold effect theory” was suggested by Battagliaet al.59 They proposed that motor neurones are exquisitely dependent upon SMN expression, requiring high amounts of the SMN protein to survive. When the required amount of the SMN protein drops below the threshold critical for the survival of motor neurones, this might lead to their degeneration and to dysregulation of apoptosis. However, reduced levels might still be compatible with the survival of other less susceptible cell types in the CNS or other tissues.
An alternative explanation was offered by Mattaj60 based on the observation of Liu et al 56 that SMN protein binds to the Sm domain in most common proteins in the U snRNP family proteins. Two of these Sm proteins are replaced in neurones by a neurone specific Sm protein (named SmN protein). Mattaj60 suggested that this form may have a different interaction with SMN thus accounting for the tissue specificity of the phenotype.
A third possibility comes from the study by Iwahashiet al,61 which helps shed some light on how lack of SMN results in the degeneration of the anterior horn cells of the spinal cord. In investigating the anti-apoptotic function of Bcl-2 (anti-apoptotic protein), a yeast two hybrid screen yielded SMN as an interactive protein. SMN on its own was shown to have a weak anti-apoptotic effect, but when coexpressed with Bcl-2 a synergistic preventative effect was observed. Furthermore, they also showed that SMN protein carrying a missense mutation Y272C17 exerts no synergism with Bcl-2. Bcl-2 coupled with SMN was shown to inhibit Bax (pro-apoptotic protein) induced apoptosis. This is particularly interesting as Bax is necessary for the death of sympathetic neurones and motor neurones.
WHAT DOES THE SMNC PROTEIN DO?
Both forms of the SMN gene encode protein with no altered amino acid sequence.17 Coovert et al 55 indicated that the SMNC protein level and number of gems differed in SMA type I from SMA type II patients, even if they possessed apparently the same number of SMNC copies. These data favour the view that the SMNC protein is not functionally equivalent. Coovertet al 55 suggested that the SMNC loci produced by conversion of the SMNTgene generates reduced but still sufficient levels of protein for gem formation (SMA type II, III), while the SMNC gene remaining in the SMA type I patient is not capable of forming gems.
Lefebvre et al 58postulated that the hyperexpression of the SMNC gene(s), therefore, can be considered as an attractive strategy for a therapeutic approach to SMA treatment. Attempts to upregulate alternative forms of a gene (for example, the γ globin gene) to overcome a genetic defect have been considered for β thalassaemia, where continued production of the fetal haemoglobin may overcome the lack of the β globin chain. The same strategy has also been proposed for Duchenne muscular dystrophy, where utrophin can substitute for dystrophin and, therefore, upregulation of the utrophin may ameliorate the clinical phenotype.62 Further studies focusing on the characterisation of the SMN promoter region63 would be essential to pursue this strategy.
THE ROLE OF NAIP
In 1996, Liston et al 25 proved that NAIP, and possibly other members of the IAP (inhibitor of apoptosis proteins) family of proteins, have anti-apoptotic effects in mammalian cells. They also showed that NAIP is expressed in motor but not in sensory neurones. This finding is in accordance with the fact that the protein is acting as a negative regulator of motor neurone apoptosis and once it is deficient or absent, it contributes to the development of the SMA phenotype. Whether NAIP is acting solely downstream on the pathological pathway of SMA or is interacting with the SMN protein, as was indicated from the two hybrid study carried out by Lu et al,64 is too early to say. Further studies are required to elucidate possible functional interactions between these proteins.
Xu et al 65 showed that overexpression of NAIP reduces the effect of transient ischaemia in rat hippocampal neurones. This opens up the exciting possibility that upregulation of NAIP expression may be used in therapy of stroke. Thus, research into a relatively uncommon disease such as SMA may offer new insights for therapy in more common disorders.
Conclusion
In conclusion, we have reviewed the currently available publications in order to develop a better understanding of the molecular basis of SMA. The clinical heterogeneity of this disorder (varying age of onset and severity of manifestation) undoubtedly mirrors the intrinsic complexity of the genes involved in its aetiology. The identification of two major genes (the survival motor neurone gene and neuronal apoptosis inhibitory protein gene) causing the SMA phenotype have provided new insight into the mechanism(s) involved in the development of this deleterious condition but highlights that the interactions between these proteins are complex and not fully resolved.
The message is therefore clear that even disorders that appear to follow simple Mendelian genetics can produce many novel and fascinating findings that continually challenge the minds of the researchers trying to untangle the knot.
Note added in proof
Scharf et al 66 have recently identified a candidate phenotypic modifier for SMA called H4F5.
Acknowledgments
We wish to thank Dr Stephen Kahler for helpful discussions and for critical reading of the manuscript.