Article Text

Abstract
Background and aims: The hepatic peptide hormone hepcidin, which has recently been isolated from human plasma and urine, is thought to be a central regulator of iron homeostasis. We investigated the presence and cellular localisation of hepcidin in the liver and developed a non-invasive assay to analyse its regulation in patients with hereditary haemochromatosis (HH), chronic renal insufficiency (CRI), and renal anaemia (RA).
Methods: Expression and localisation of hepcidin was shown by reverse transcription-polymerase chain reaction, western blot, immunocytochemistry, and immunofluorescence in human and guinea pig liver. Serum concentrations were determined in various groups of patients using a sensitive enzyme linked immunosorbent assay (ELISA).
Results: Western blot analysis with region specific antibodies identified a ~10 kDa peptide corresponding to the apparent molecular mass of pro-hepcidin. Localisation studies revealed that pro-hepcidin is expressed at the basolateral membrane domain of hepatocytes and is also present in blood. We developed a stable sensitive ELISA for detection and determination of pro-hepcidin in human serum. Mean pro-hepcidin level in human serum of healthy volunteers was 106.2 ng/ml. Enhanced levels of pro-hepcidin (148.1 ng/ml) were found in patients with CRI but normal haemoglobin values, indicating that the kidneys may metabolise and/or eliminate the circulating hormone. In contrast, concentrations of pro-hepcidin were significantly decreased in patients with HH (70.2 ng/ml) and also in patients with RA (115.0 ng/ml) compared with the CRI group.
Conclusions: From the detection of pro-hepcidin in human serum, we conclude that the prohormone may be involved in the regulation of iron metabolism in HH. Decreased pro-hepcidin levels could play an important role in the pathogenesis of HH.
- hepcidin
- chronic renal insufficiency
- iron absorption
- hereditary haemochromatosis
- liver
- aa, amino acid
- CRI, chronic renal insufficiency
- ELISA, enzyme linked immunosorbent assay
- EPO, erythropoietin
- HH, hereditary haemochromatosis
- PBS, phosphate buffered saline
- RA, renal anaemia
- RT-PCR, reverse transcription-polymerase chain reaction
- TfR1, classical transferrin receptor
- TfR2, transferrin receptor type 2
- TBS, Tris buffered saline
Statistics from Altmetric.com
- aa, amino acid
- CRI, chronic renal insufficiency
- ELISA, enzyme linked immunosorbent assay
- EPO, erythropoietin
- HH, hereditary haemochromatosis
- PBS, phosphate buffered saline
- RA, renal anaemia
- RT-PCR, reverse transcription-polymerase chain reaction
- TfR1, classical transferrin receptor
- TfR2, transferrin receptor type 2
- TBS, Tris buffered saline
Iron is an essential trace element that is required for growth and development of all living organisms; it is indispensable for DNA synthesis and a broad range of metabolic processes. However, disturbances in iron metabolism are implicated in a number of significant human diseases, including iron deficiency anaemia, haemosiderosis, or the iron overload disease haemochromatosis.1–3 Under physiological conditions, the body’s iron content is regulated by controlling absorption. In mammals, iron absorption occurs predominantly in the duodenum and upper jejunum, and is the only mechanism by which iron stores are physiologically controlled.2 Following absorption, iron is bound to circulating transferrin and delivered to tissues throughout the body. In the liver, the major site of iron storage, transferrin bound iron is taken into the cells by receptor mediated endocytosis via the classical transferrin receptor (TfR1)4 and presumably in greater amounts via the recently identified homologous transferrin receptor 2 (TfR2).5
A feedback mechanism exists that enhances iron absorption in individuals who are iron deficient whereas iron absorption is reduced in those with iron overload.1–3 In hereditary haemochromatosis (HH) however, this regulatory mechanism seems to be impaired; despite iron overload, elevated amounts of iron are absorbed from the diet and lead to accumulation of excess iron in internal organs, resulting in organ dysfunction and failure. The molecular mechanisms by which the intestine responds to alterations in body iron requirements is poorly understood. In this context, hepcidin, a recently identified mammalian peptide,6,7 is predicted as a key signalling component regulating iron homeostasis.2,8
Hepcidin was initially isolated as a 25 amino acid (aa) peptide in human plasma and urine, exhibiting antimicrobial activity.6,7 A hepcidin cDNA encoding an 83 aa precursor in mice and an 84 aa precursor in rats and humans, including a putative 24 aa signal peptide, was subsequently identified in the search for liver specific genes that were regulated by iron.9 Hepcidin expression is abolished in mice exhibiting iron overload due to targeted disruption of the upstream stimulatory factor 2 (Usf2) gene, resembling the same phenotype as found in hfe−/− mice.10 Hence, this indicates that this peptide plays a pivotal role in iron metabolism. In contrast, overexpression of hepcidin was shown to result in severe iron deficiency anaemia in transgenic mice,8 indicating that hepcidin is a central regulator of iron homeostasis. Moreover, recent studies have shown that liver hepcidin expression is decreased in the hfe knockout mouse,11 and mutations in the hepcidin peptide are associated with severe juvenile haemochromatosis,12 providing new perspectives in our understanding of the molecular pathogenesis of iron overload. However, the mechanism by which hepcidin balances the body’s iron stores or adjusts dietary iron absorption under physiological and pathological conditions still remains to be identified.
In this respect, cellular localisation of this peptide and its regulation in various iron states are of major importance in the study of hepcidin function. Although northern blot analysis of human and mouse hepcidin mRNA levels in various organs revealed that hepcidin is predominantly expressed in the liver,6–8 no data exist on the cellular localisation of this peptide. To gain a better understanding of the role of hepcidin under physiological conditions and in relevant diseases, we generated specific antibodies against the midportion and the C terminus of the hepcidin precursor molecule. These antibodies allowed us to define the cellular localisation of hepcidin in the human and guinea pig liver. We established a sensitive enzyme linked immunosorbent assay (ELISA) enabling us to detect pro-hepcidin in the human serum of patients with HH, chronic renal insufficiency (CRI), and renal anaemia (RA). Our results suggest that pro-hepcidin is released across the hepatocyte basolateral membrane into the blood and is subjected to renal elimination. As serum levels of hepcidin are remarkably downregulated in HH and chronic RA, we assume that hepcidin plays a role in the pathophysiology of these diseases.
MATERIALS AND METHODS
Tissues and tissue preparation
Human liver samples (n = 7) were obtained after hemi-hepatectomy in adult patients with liver metastases. Healthy tissues were fixed in 4% paraformaldehyde for immunohistochemistry or immediately frozen in liquid nitrogen for reverse transcription-polymerase chain reaction (RT-PCR), western blot, and immunofluorescence analysis.
Guinea pigs (n = 7) were anaesthetised and subsequently sacrificed by cervical dislocation. Tissue specimens from liver and skeletal muscle were resected and immediately frozen in liquid nitrogen for western blot analysis or fixed in paraformaldehyde.
Peptide synthesis, immunisation procedure, and antibodies
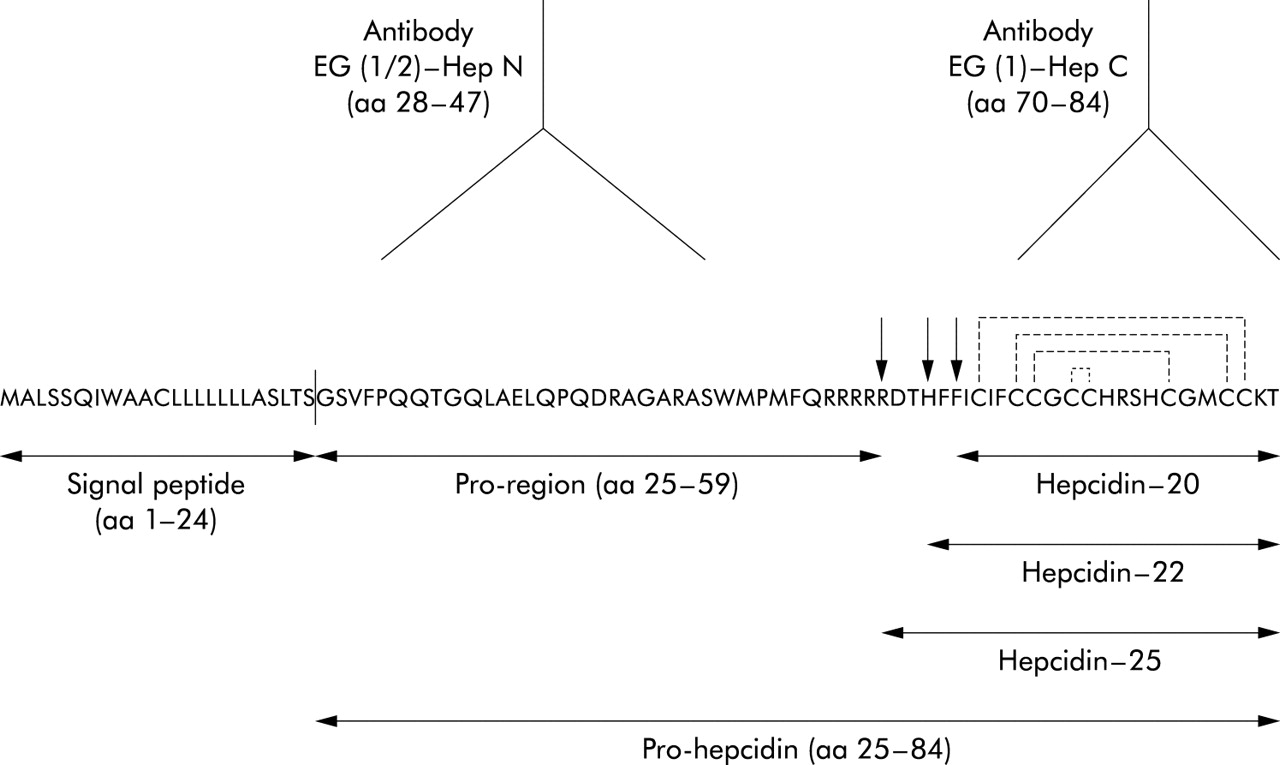
From the published pro-hepcidin sequence,6,9 the peptides hepcidin-(28–47) and hepcidin-(70–84) were synthesised as C terminal amides using a standard Fmoc protocol.13 Peptides were coupled to keyhole limpet haemocyanin using m-maleimidobenzoyl-N-hydroxysuccinimide ester, and two SPF rabbits (Charles River-Iffa Credo Eurogentec, Seraing, Belgium) were immunised with each peptide conjugate (Eurogentec, Seraing, Belgium). After testing the titre by ELISA, three antisera (EG(1)-HepC directed against hepcidin-(70–84) and EG(1)-HepN and EG(2)-HepN, each directed against hepcidin-(28–47)) were used in the present study (fig 1). The peptide epitopes used for generation of the antisera displayed no homology to any hitherto reported protein, as confirmed by the BLAST P2 search.

Amino acid sequence of the human hepcidin precursor protein containing a typical 24 amino acid (aa) signal peptide at the N terminus (the line between aa 24 and 25 indicates the putative signal sequence cleavage site), a 35 aa pro-region, and the C terminal 20-, 22-, and 25-aa hepcidin peptides differing only by their N terminal truncation, as denoted by the arrows. After cleavage of the signal peptide from hepcidin precursor, the pro-hepcidin molecule is produced consisting by 60 aa. The proposed disulphide connectivity in hepcidin 25 is 1–8, 2–7, 3–6, and 4–5, as shown by the broken lines.20 The antisera EG (1 and 2)-Hep N are raised against hepcidin precursor aa 28–47 while the antiserum EG(1)-HepC is raised against aa 70–84, as denoted by the antibody symbols.
As peptide antisera generally work cross species because of sequence homology between species, the hepcidin antisera, raised against human hepcidin, were also used in guinea pig tissues.
Expression analyses in the human liver
RNA isolation was performed using Qiagen RNAeasy kit, including DNA digestion. RT-PCR analysis was performed as described previously14,15 using the following primers and specifications given in 5′–3′ orientation: human hepcidin (GenBank database accession No NM021175), 5′-CTG CAA CCC CAG GAC AGA G-3′ and 5′-GGA ATA AAT AAG GAA GGG AGG GG-3′; corresponding to positions 147–165 and 338–316. After initial denaturation of 94°C for four minutes, reactions were subjected to 35 cycles of the following thermal programme: 94°C for 30 seconds, 60°C for 30 seconds, and 72°C for 30 seconds; this program was followed by a final five minute elongation step at 72°C. Amplification products were run on an ethidium bromide stained 1.8% 89 mM Tris/89 mM boric acid/2 mM EDTA (pH 8.3) agarose gel. Amplification of significant levels of genomic DNA was excluded by appropriate controls.
Expression analyses in HepG2 cells
Human hepatoma HepG2 cells were obtained from the German Collection of Microorganisms and Cell Culture (Braunschweig, Germany) and grown at 37°C in 5% CO2 in RPMI 1640 media (Gibco, Karlsruhe, Germany) supplemented with 10% (vol/vol) heat inactivated fetal bovine serum, penicillin (100 units/ml), and streptomycin (100 µg/ml). Cells were analysed by RT-PCR using the primer specifications described above. For immunofluorescence microscopy, HepG2 cells were grown on glass slides, fixed for four minutes in methanol, and permeabilised with 0.5% Triton X-100 in phosphate buffered saline (PBS). After incubation with hepcidin antibodies (1:2000) for 60 minutes, followed by incubation with Cy-3-conjugated antirabbit antibody (Dianova, Hamburg, Germany), immunostaining was investigated under an Olympus AX70 microscope using appropriate filters.
Extraction of hepcidin from serum, tissues, and HepG2 cells
As a larger source of hepcidin, we used serum collected from patients with chronic renal failure. For extraction of hepcidin, 20 ml serum samples were diluted 1:1 with 0.01 N HCl and adjusted to pH 3.0 with concentrated HCl. Frozen tissues and HepG2 cells were mixed in 0.5 M acetic acid and boiled for eight minutes, as described previously.13,16 After homogenisation with an Ultra-Turrax homogeniser (Janke and Kunkel, Staufen, Germany), samples were centrifuged at 20 000 g for 20 minutes at 4°C and the supernatants were filtered through a 0.45 µm pore size filter. To enrich proteins, serum samples, cell, and total tissue extracts were applied to an octadecasilyl (C18) Sep-Pak cartridge (Waters, Massachusetts, USA). The column was washed with 0.01 M HCl and eluted with 30% (vol/vol) 2-propanol/30% (vol/vol) methanol/0.01 M HCl.13 Protein fractions were lyophilised and stored at −80°C until use.
Immunoblot analysis
For western blot analysis, protein extracts were incubated for seven minutes at 94°C in sample buffer with 4% (wt/vol) sodium dodecyl sulphate (Merck, Darmstadt, Germany), 50 mM Tris HCl (pH 8.45), 1 mM EDTA, 3.24 mM dithiothreitol (Roth, Karlsruhe, Germany), 12.5% (wt/vol) glycerol (Merck), and 0.002% bromophenol blue (Merck). To detect hepcidin, a 16.5% tricine-sodium dodecyl sulphate-polyacrylamide gel was used according to published protocols.13–16 Following electrophoresis, proteins were transferred onto hydrophobic polyvinylidene fluoride based membranes (Pall, Portsmouth, UK) by semi dry blotting. Membranes were incubated overnight with hepcidin antibodies at the dilutions indicated above. After washing in Tris buffered saline containing 10 mM Tris HCl (pH 8.0), 150 mM NaCl, and 0.05% Tween 20, immunoreactive proteins were visualised after incubation with alkaline phosphatase conjugated goat antirabbit antibody (diluted 1:50 000; Sigma, St Louis, Missouri, USA) using nitro blue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate as chromogens (Sigma). The immunoreaction on the western blot was specifically blocked after preincubation of the antibodies with the corresponding peptide immunogens. Cross reactivity with the second goat antirabbit antibody was excluded by appropriate controls.13–16
Immunohistochemistry and immunofluorescence
Tissues were fixed in 4% paraformaldehyde for 18 hours at 4°C. After dehydration in graded ethanol series, specimens were embedded in paraffin. Paraffin sections (5 µm) were immunostained for hepcidin (antibodies EG(1)-HepN, EG(2)-HepN, and EG(1)-HepC, each diluted 1:2000) by the avidin-biotin-peroxidase complex technique and incubation sequences, as previously described.14,15 Sections were incubated with the respective antibodies for 24 hours at 4°C, followed by incubation with biotinylated antirabbit IgG (Jackson Immunoresearch, West Grove, Pennsylvania, USA) for 30 minutes diluted 1:200. Sections were then incubated for 30 minutes with a preformed complex of biotin-peroxidase/streptavidin (Jackson Immunoresearch), diluted in PBS (final concentrations: biotin-peroxidase 0.7 µg/ml; streptavidin 5 µg/ml). Antigen-antibody binding sites were visualised by incubation of the sections in 0.7 mM diaminobenzidine hydrochloride/0.002% H2O2 in 0.05 M Tris HCl (pH 7.6).
For immunofluorescence microscopy, tissue sections from human liver (2–4 µm) were prepared with a cryotome (FrigoCut 2800E; Leica, Nussloch, Germany), air dried for two hours, and fixed for 10 minutes in cold acetone (−20°C). Double immunofluorescence labelling was performed as described previously17 using the specific hepcidin antibodies (diluted 1:1000) and monoclonal antibody C21917 raised against canalicular P-glycoproteins (Centocor, Malvern, Pennsylvania, USA) diluted 1:30. After incubation with the respective antisera, staining was performed by incubation with Cy2- (1:200) and Cy3- (1:600) labelled antibodies against mouse and rabbit IgG (Dianova, Hamburg, Germany). Micrographs were taken with an Olympus AX70 microscope equipped with a digital camera (colour view 12, soft imaging system SIS, Münster, Germany) and analySIS software (SIS, Münster, Germany).
Specificity controls
Method dependent non-specificities were excluded by running controls, as described previously.13,16 Antibody specificities were tested by preadsorption of antibodies with homologous and heterologous antigenic peptides (6.25–100 µg/ml of the antiserum).14,15 Preadsorption of antibodies with homologous antigens at concentrations as low as 6.25 µg/ml completely blocked immunostaining in liver tissues and cells while preadsorption of antibodies with heterologous antigens at concentrations up to 100 µg/ml had no effect on immunostaining.
Hepcidin ELISA competitive binding assay
Serum samples were obtained from 26 healthy individuals (13 women, 13 men; aged 26–64 years, mean 43), 35 patients with HH homozygous for the C282Y mutation in HFE (14 women, 21 men; aged 23–82 years, mean 54), with (15 patients) and without (20 patients) blood letting therapy, and 59 patients with renal insufficiency undergoing chronic haemodialysis (33 women, 26 men; aged 26–96 years, mean 57). During sample collection we ensured that patients had no infection. Nineteen patients in the renal insufficiency group had renal anaemia, characterised by a maximum haemoglobin concentration of 11 g/dl. All patients with CRI were treated 2–3 times a week with 3000 IE recombinant human erythropoietin (EPO). Blood samples (10 ml) were withdrawn into serum tubes and centrifuged at 2500 g for 10 minutes at 4°C. Determinations were performed in duplicate using 96 well microtitre plates coated with 200 µl/well rabbit antihepcidin antibody EG(2)-HepN diluted 1:4000 in Tris buffered saline (TBS) containing 40 mM Tris HCl (pH 7.3), 100 mM NaCl. Standards (50 µl) containing various amounts of synthetic peptides (0, 20, 100, 500, and 1000 ng/ml) or human serum samples and 150 µl N terminally biotinylated hepcidin-(28–47) (Peptide Specialty Laboratories GmbH, Heidelberg, Germany) (2 ng/well) were added to each well and incubated for one hour at room temperature. After washing with TBST (TBS with 0.05% Tween 20), biotinylated antigen-antibody complexes were detected by streptavidin-peroxidase enzyme (Dako, Hamburg, Germany) with the substrate tetramethylbenzidine (DRG Instruments GmbH, Marburg, Germany); the colour reaction was stopped with 1 M H2SO4 and extinction of the solution was read at 450/630 nm wavelength.
ELISA characterisation
Details of ELISA characterisation are available from the authors on request.
Data management and statistics
Measured values of hepcidin in the four groups were entered in an EXCEL spreadsheet and evaluated using SAS WIN version 8.2. Measured values were summarised by means of the following summary statistics by diagnosis group: number of observations, arithmetic mean, SD, minimum, median, and maximum. Possible differences between groups were analysed with pairwise Wilcoxon U tests. The level of significance α was set at 5% (0.05).
Correlation between pro-hepcidin and iron, ferritin, or transferrin was analysed using the Spearman rank correlation.
RESULTS
Expression of hepcidin in the liver and HepG2 cells
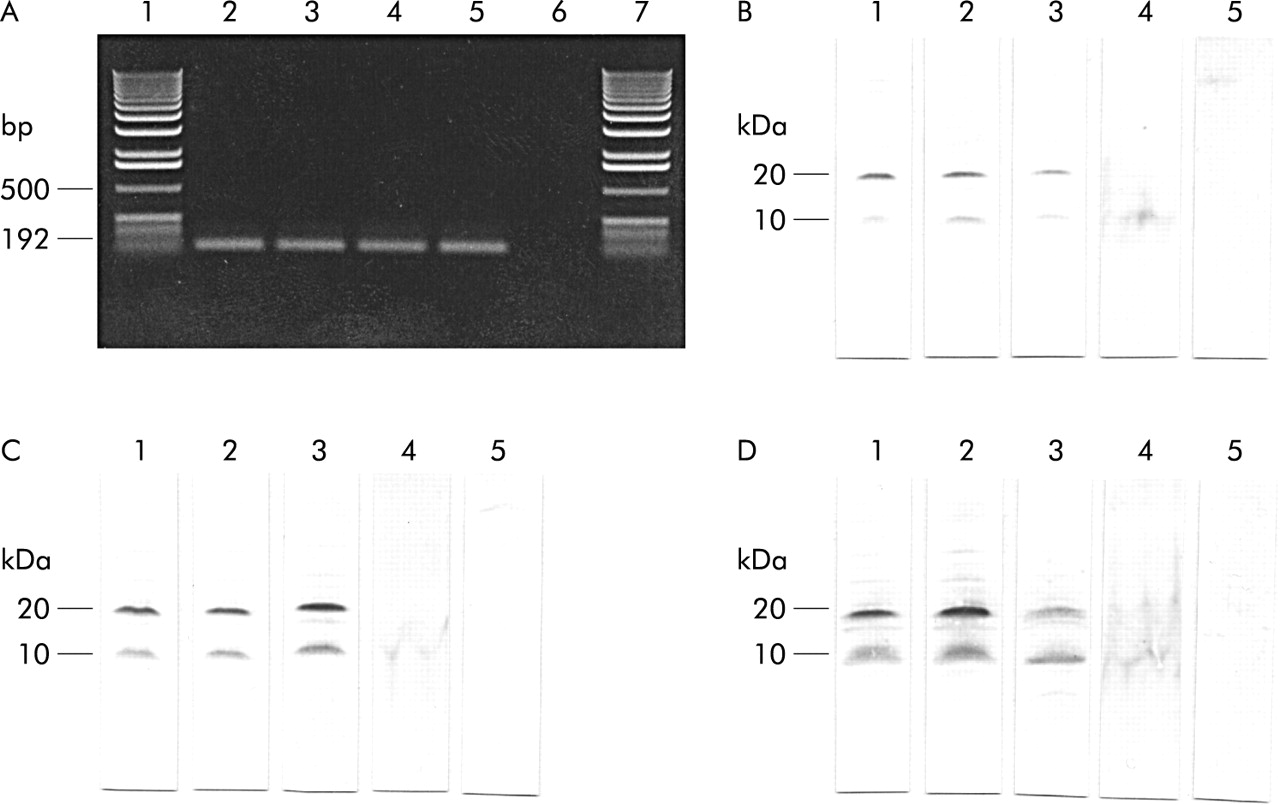
RT-PCR analysis demonstrated that hepcidin is expressed in human liver.18 Similarly, a 192 bp expected PCR product was detected in HepG2 cells (control) which were already shown to express hepcidin (fig 2A).9,18 In western blot analysis, all hepcidin antibodies (EG(1)-HepN, EG(2)-HepN, and EG(1)-HepC) coincidentally identified an immunoreactive band of ~10 kDa in extracts of human and guinea pig liver. This liver peptide co-migrated with an immunoreactive band recognised by the hepcidin antibodies in homogenates of HepG2 cells (fig 2B–D). All antibodies also identified an immunoreactive protein at ~20 kDa in all lanes loaded with human and guinea pig liver extracts or HepG2 cell extracts. Western blot analysis of skeletal muscle extracts (control) showed neither the immunoreactive band of 10 kDa nor the band at 20 kDa (fig 2B–D).

(A) Reverse transcription-polymerase chain reaction analysis of human liver (lanes 2 and 3) and HepG2 cells (lanes 4 and 5) showing gene expression of hepcidin. A bp DNA ladder is indicated (lanes 1 and 7). Lane 6 show a negative control. (B–D) Western blot analyses of hepcidin in extracts of guinea pig (lane 1) and human liver (lane 2) as well as in HepG2 cells (lane 3), human serum (lane 4), and guinea pig skeletal muscle (lane 5, control) with antibodies EG(1)-HepN (B), EG(2)-HepN (C), and EG(1)-HepC (D). Note the immunoreactive bands at 10 and 20 kDa obtained with all antibodies recognising different epitopes in the hepcidin precursor. (Molecular mass markers used: phosphorylase B 105 kDa; glutamic dehydrogenase 53 kDa; carbonic anhydrase 34 kDa; myoglobin blue 23 kDa; myoglobin red 17 kDa; lysozyme 13 kDa; aprotinin 7 kDa; and insulin 3 kDa.)
Immunofluorescence in HepG2 cells
Using epitope specific antihepcidin antibodies, expression of hepcidin peptide in HepG2 cells was investigated by immunofluorescence analysis. All antibodies similarly identified hepcidin in HepG2 cells resulting in strong immunoreactivity (fig 3).

Detection of hepcidin in HepG2 cells by immunofluorescence microscopy using the antibodies EG(1)-HepN (A), EG(2)-HepN (B), and EG(1)-HepC (C) (scale bar 8 µm).
Cellular and subcellular localisation of hepcidin
Immunohistochemical studies with various region specific antibodies consistently localised hepcidin to hepatocytes in human liver (fig 4). Kupffer cells, endothelial cells, bile ducts, and the vascular system completely lacked hepcidin immunoreactivity. The same antibodies detected strong hepcidin immunoreactivity in guinea pig liver also (fig 4). Hepatic lobules were heterogeneous with respect to hepcidin immunoreactivity: within a hepatic lobule, hepcidin immunoreactive cells were predominantly located in periportal zones, and the frequency of hepcidin positive cells continuously decreased from the portal triads towards the central veins (fig 5). Notably, distinct intercellular differences existed between hepcidin positive cells: while most hepatocytes were strongly positive for hepcidin others displayed only faint staining or were totally unreactive for hepcidin (fig 5).

Cellular localisation of hepcidin in guinea pig (A–F) and human (G–I) liver. The paraffin sections immunostained with the region specific antibodies EG(1)-HepN (A, D, G), EG(2)-HepN (B, E, H), and EG(1)-HepC (C, F, I) showed distinct immunoreactivity at the basolateral membrane domain of hepatocytes (arrows). (Magnification: A–C ×180; D–I, ×540.)
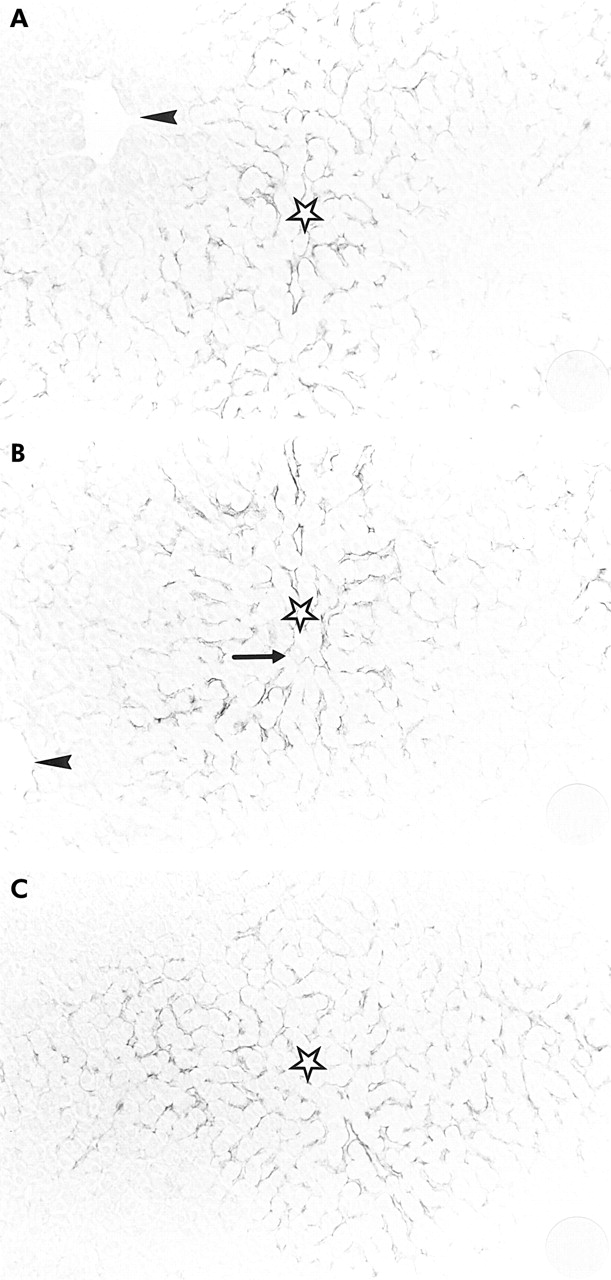
Immunohistochemical sections of guinea pig liver (A, antibody EG(1)-HepN; B, antibody EG(2)-HepN; C, antibody EG(1)-HepC) showing the clear zonation of hepcidin within hepatic lobules with decreasing immunoreactivity from periportal zones (stars) towards the central veins (arrowheads). Note that no immunoreactivity was found in hepatocytes around the central veins. (The arrow in (B) indicates a portal triad.) (Magnification: A–C ×180.)
At the subcellular level, hepcidin immunoreactivity was confined to the basolateral ( = sinusoidal) membrane domain of hepatocytes by immunohistochemistry; no immunoreactivity was found at the apical membrane domain of the respective cells (fig 4). Similarly, immunofluorescence analysis demonstrated strong immunoreactivity for hepcidin at the basolateral membrane domain; immunoreactivity was absent from the apical membrane domain as revealed by double staining with the C219 antibody raised against canalicular P-glycoproteins17 (data not shown).
Detection of hepcidin propeptide in human serum
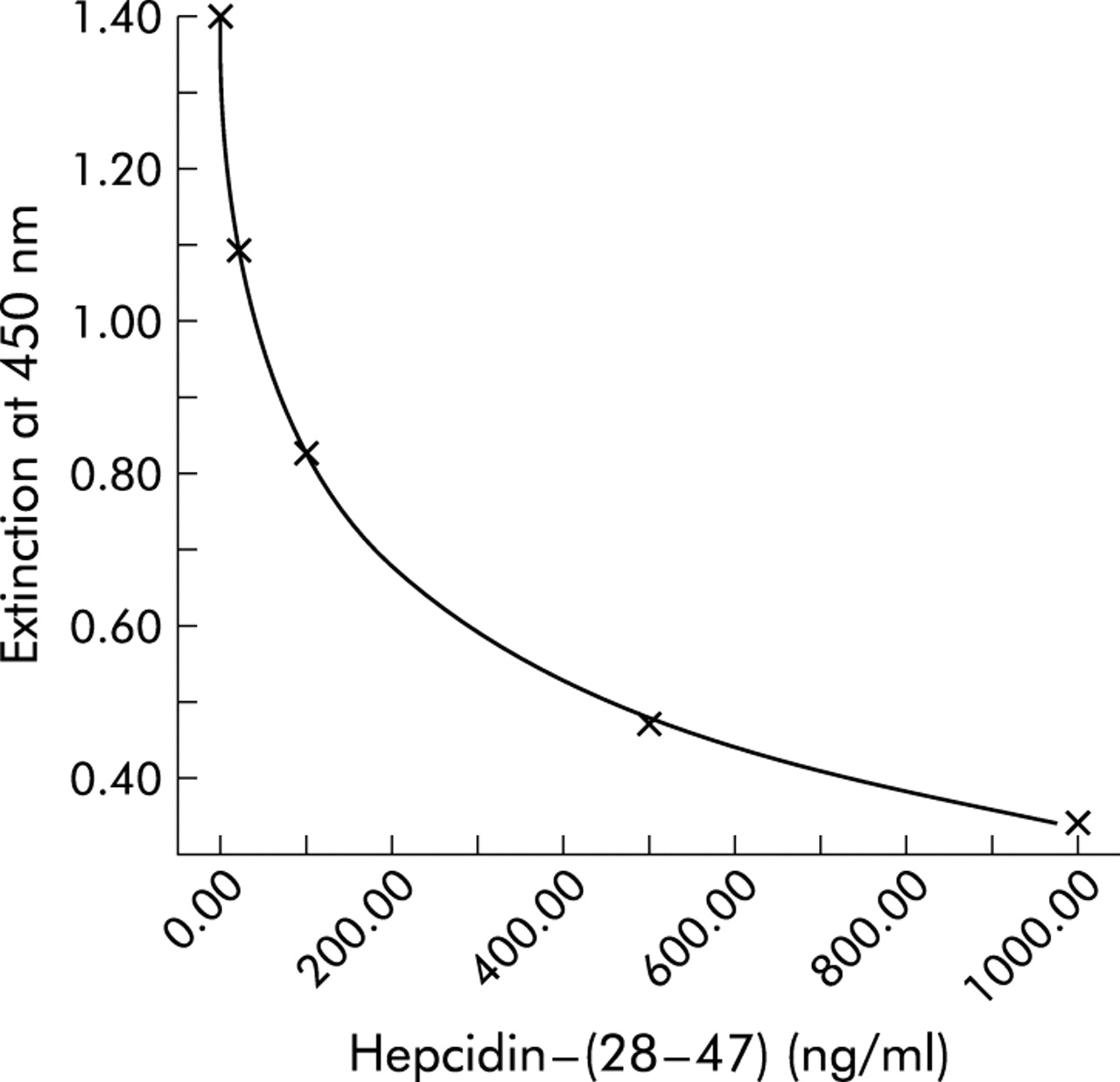
A stable pro-hepcidin ELISA assay (DRG Instruments GmbH, Marburg, Germany) with high reproducibility and sensitivity was developed with the specific N terminal hepcidin antibody EG(2)-HepN. As shown in fig 6, ELISA revealed the highest resolving power between 4 and 400 ng/ml, a range where pro-hepcidin concentrations in human serum are determined. As a specificity control, incubation in ELISA was performed with heterologous peptides. No cross reactivity was observed when heterologous peptides were used.
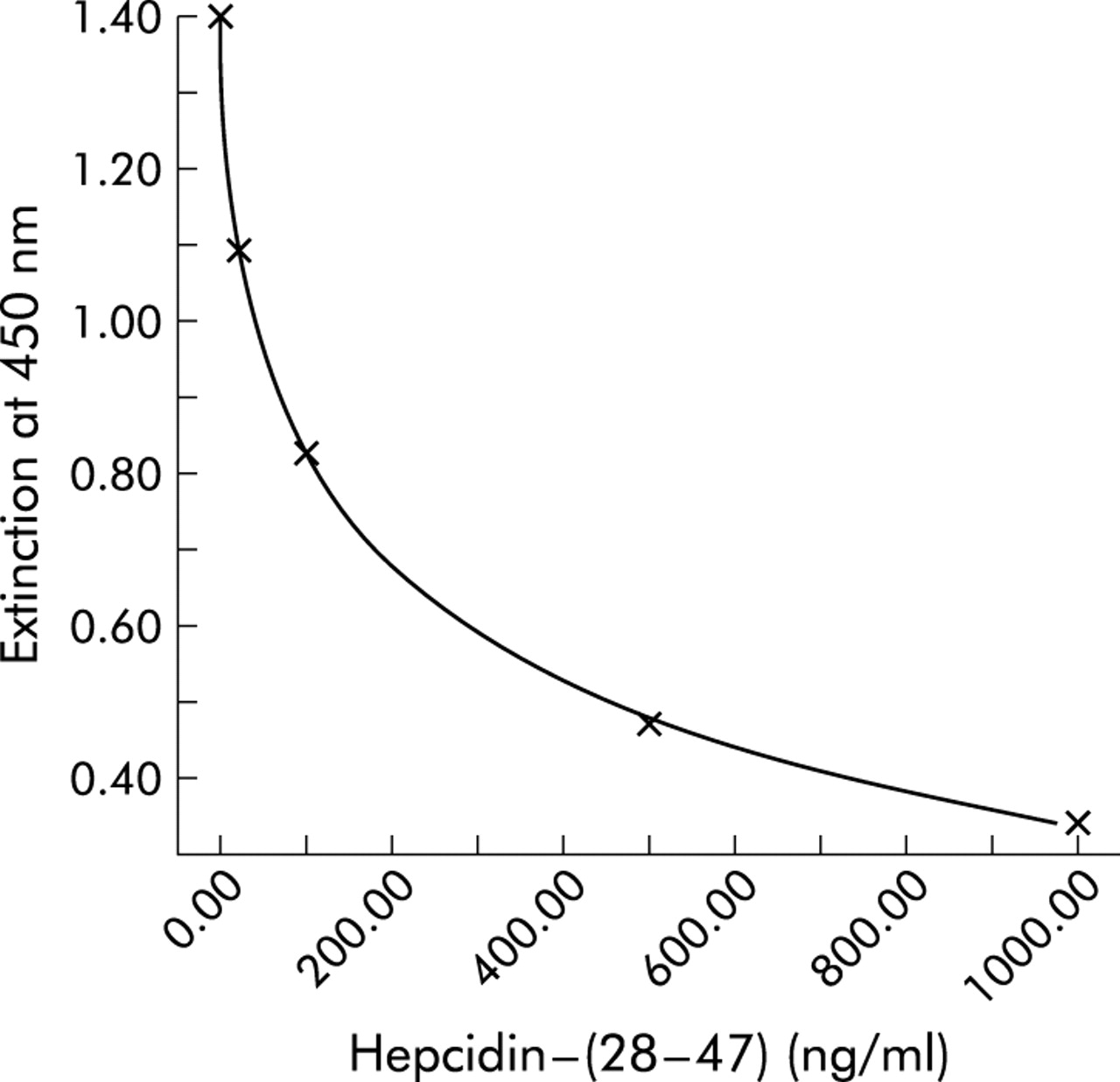
Enzyme linked immunosorbent assay (ELISA) for circulating human pro-hepcidin. A representative standard curve with concentrations of hepcidin-(28–47) and extinction of the ELISA solution at a wavelength of 450 nm are shown. Note the high resolving power in the range 4–400 ng/ml hepcidin-(28–47).
The presence of pro-hepcidin in blood was verified by western blotting analysis. In extracts of human serum, all hepcidin antibodies identified a single hepcidin immunoreactive band of ~10 kDa molecular mass that co-migrated exactly with the immunoreactive hepcidin in liver tissues and HepG2 cell extracts (fig 2B–D).
ELISA characteristics
The sensitivity of the assay was 3.95 ng/ml. There was no overlap with the lowest standard (20 ng/ml). Serial dilutions of human pro-hepcidin, dissolved in the zero standard, ran parallel to the standard curve of the pro-hepcidin ELISA, with the range of recovery between 90.6% and 111.6%. Recovery expressed as a percentage of observed from expected concentration was between 91.8% and 105.7%. Good precision was demonstrated (total coefficient of variance <10%) at three concentrations of pro-hepcidin tested across the assay range.
Pro-hepcidin levels in hereditary haemochromatosis, chronic renal insufficiency, and renal anaemia
Using the sensitive hepcidin ELISA, pro-hepcidin in the range 51.6–153.4 ng/ml serum (mean 106.2 (SEM 32.1) ng/ml) was detected in the healthy control group of 26 volunteers (fig 7, table 1). In patients with HH, concentrations of pro-hepcidin were 12.1–153.9 ng/ml serum (mean 70.2 (SEM 38.1) ng/ml). These concentrations were significantly lower compared with those in control subjects (p<0.05) (fig 7, table 1). Pro-hepcidin concentrations varied from 31. 1 to 471.3 ng/ml (mean 148.1 (SEM 88.0) ng/ml) in serum of patients suffering from CRI and were significantly increased compared with those in control subjects (p<0.01) and HH patients (p<0.001). In contrast, pro-hepcidin levels in haemodialysis patients with RA were significantly decreased (115.0 (53.1) ng/ml; range 20.5–252.4) (p = 0.05) compared with patients with CRI (fig 7, table 1).
Results of pairwise U tests (p values)

Box plot of values for venous serum pro-hepcidin concentrations in 26 healthy volunteers (control), 40 patients with chronic renal insufficiency, 19 patients with chronic renal insufficiency and renal anaemia, and 35 patients with hereditary haemochromatosis. The line within the box indicates the median, the circle indicates the mean. The lower and upper edges of the box indicates the first and third quartiles, and the whiskers the minimum and maximum values. The broken line marks the mean level of the control group for circulating immunoreactive pro-hepcidin (106.16 ng/ml).
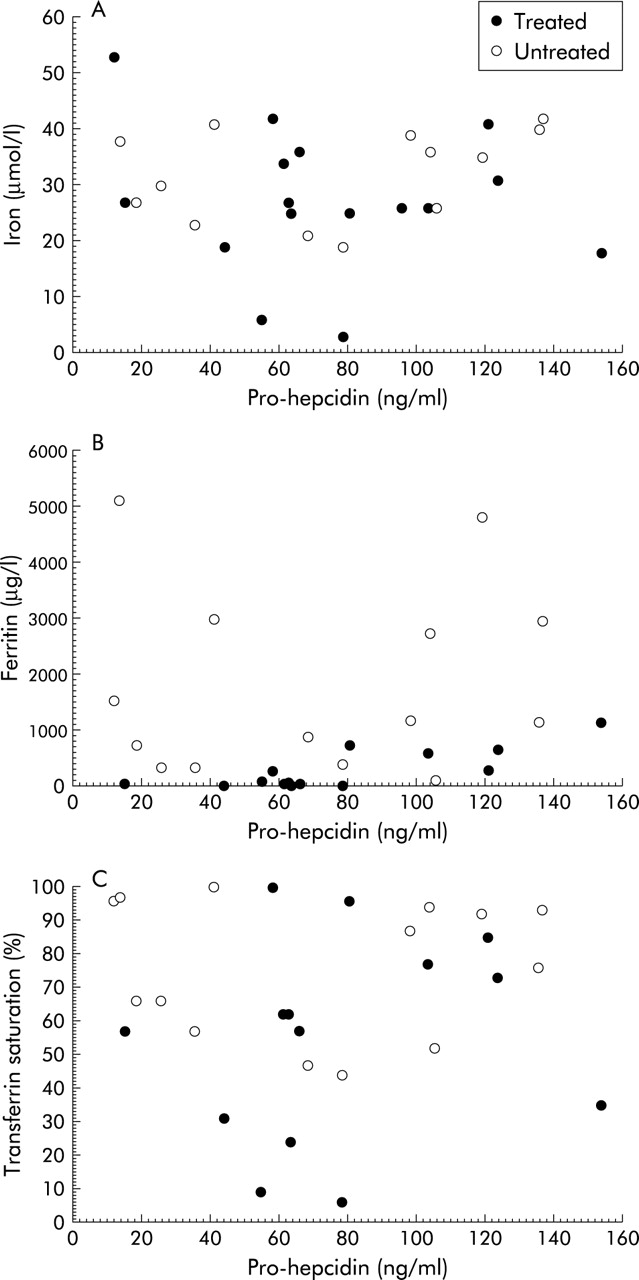
No significant correlation was found in our samples (serum from HH, CRI, and RA) between pro-hepcidin and iron, ferritin, or transferrin saturation (fig 8). The test of difference from zero was not significant.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Correlation between pro-hepcidin and iron (A), ferritin (B), and transferrin saturation (C) in samples of treated and non-treated hereditary haemochromatosis patients. Note that no remarkable correlation was found in our samples.
DISCUSSION
RT-PCR analyses with specific primers showed that hepcidin is highly expressed in HepG2 cells (control), a well differentiated hepatocellular carcinoma cell line19 demonstrating in many respects the physiology of normal hepatocytes. Using the appropriate primer specifications and combinations successfully employed in HepG2 cells, the RT-PCR studies confirmed expression of hepcidin in the human liver. Three different antibodies recognising different epitopes in the hepcidin precursor molecule (fig 1) concurrently identified an immunoreactive peptide of ~10 kDa by western blot analysis not only in HepG2 cells but also in liver extracts of two species, humans and guinea pig. The apparent molecular mass of this immunoreactive peptide is in accordance with the predicted molecular mass deduced for the hepcidin prohormone from the cDNA sequence (fig 1).9 Interestingly, a second immunoreactive band of ~20 kDa was detected by all hepcidin antibodies in extracts of HepG2 cells and human and guinea pig liver but was absent in control tissue. This immunoreactive protein may reflect a dimeric type of hepcidin. In fact, in a previous study, an aggregation property and possible formation of multimers were described for hepcidin-25 but not for hepcidin-20.20
Immunocytochemical studies with the region and molecular domain specific hepcidin antibodies revealed strong immunoreactivity in HepG2 cells, demonstrating expression of hepcidin in these cells, as already shown using molecular biological techniques.18 Immunohistochemical and immunofluorescence investigations with these different hepcidin antibodies indicated that, in human and guinea pig liver, hepcidin is specifically localised in hepatocytes mainly located around the portal triads. Coincident staining by different region specific antibodies not only in human and guinea pig liver but also in HepG2 cells indicates hepatocytes as the source of hepcidin. Hepcidin immunoreactivity decreased from the periportal zones towards the central veins. This zonation within the portal lobules may have a functional significance as periportal hepatocytes have first pass access to portal veins, bringing iron rich blood from the gut. Notably, distinct intercellular differences existed between hepcidin positive cells with respect to the density of hepcidin immunoreactivity that may reflect intercellular differences in expression or secretion of hepcidin.
At the subcellular level, hepcidin was concentrated at the basolateral membrane domain of hepatocytes. No immunoreactivity was found at the apical membrane domain. The discrete distribution pattern of hepcidin at the subcellular level may infer basolaterally directed release of hepcidin into the liver sinusoids. This directional secretion route is additionally substantiated by detection of hepcidin prohormone (fig 1) in human serum (see below); consequently, these findings provide further evidence that hepatocytes may regulate iron metabolism in an endocrine fashion via secretion of pro-hepcidin.
As blood forming tissues and sites of iron storage, such as the liver, are thought to transmit signals to intestinal cells that indicate the body’s requirements for dietary iron,2 we presume that hepcidin is a candidate signalling factor secreted from hepatocytes and regulating intestinal iron absorption. However, there is still controversy concerning the existence of certain molecular forms of hepcidin in blood.6,7,20
To analyse whether the prohormone of hepcidin is secreted into blood, and to assess the range of pro-hepcidin levels in human serum of healthy volunteers and patients with different diseases, an ELISA was developed by applying the N terminal antibody EG(2)-HepN raised against hepcidin prohormone. Although the C terminal antibody EG(1)-HepC revealed specific results in dot blot (data not shown), western blot, immunohistochemistry, and immunofluorescence experiments (figs 1–4), no immunoreactivity was obtained in ELISA experiments. The compact folding pattern of hepcidin and its tertiary structure in blood may account for the inability of the EG(1)-HepC antibody to identify circulating hepcidin.
ELISA with antibody EG(2)-HepN was characterised by high reproducibility, stability, and sensitivity, with a detection limit of 3.95 ng/well and a powerful resolution in the range 4–400 ng/ml; the range where hepcidin concentrations were determined. In human serum from healthy individuals (n = 26), pro-hepcidin was measured in the range 51.6–153.4 ng/ml (mean 106.2 (SEM 32.1) ng/ml), which is comparable with the concentration of known regulating peptide hormones and approximately 11-fold higher than the concentration of hepcidin in human urine.7 Interestingly, measured concentrations exhibited a wide range of pro-hepcidin, indicating that the peptide may be subject to strong regulation.
The cDNA structure suggests that hepcidin is translated as an 84 aa pre-propeptide that is N terminally processed to the 20–25 aa peptides7 (fig 1). Although a strong consensus sequence for a signal sequence cleavage site is located between Gly24 and Ser25 that would result in a 60 residue propeptide, previous studies failed to isolate the larger propeptide from native sources such as liver tissue and blood.7 In addition to technical difficulties, the abundance of propeptide convertases in the liver may inhibit isolation of certain propeptides. In this context, recent studies have shown that the human circulating form of hepcidin, described by two research groups in blood6 and in urine,7 consists of the C terminal 20–25 aa of the protein. However, our ELISA measurements were performed with the specific antibody raised against the N terminus of hepcidin precursor, implying that apart from the 20–25 aa processed forms, hepcidin prohormone is secreted and circulates in human blood. Indeed, the potential release of pro-hepcidin into blood was confirmed by western blot analysis. In extracts of human serum, all hepcidin antibodies identified a single hepcidin band of ~10 kDa that co-migrated exactly with the immunoreactive hepcidin in tissue extracts of the liver and HepG2 cells (positive control; fig 1). Hepcidin fragments smaller than 10 kDa were not detected. The presence of pro-hepcidin in human serum suggests that hepatocytes secrete the prohormone of hepcidin that may decrease dietary iron absorption via an endocrine pathway.
To analyse the significance of hepcidin in patients with iron overload, we determined hepcidin concentrations in the serum of 35 HH patients homozygous for the C282Y mutation in HFE, with typical characteristics of iron overload detected in all HH patients studied. Hepcidin concentrations were not increased in these individuals, to reduce intestinal iron absorption, as thought previously.21 Pro-hepcidin levels in the serum of HH patients were unexpectedly downregulated, not only in untreated patients but also in individuals undergoing weekly blood letting therapy. Compared with healthy volunteers, mean pro-hepcidin concentrations were markedly reduced from 106.2 to 70.2 ng/ml serum. No difference was observed between treated and untreated HH patients. These findings are in line with previous HH studies showing that liver hepcidin expression is significantly decreased in the hfe knockout mouse11,22 and in patients with HFE associated haemochromatosis.23 They are also in accordance with in vitro studies demonstrating that iron loading of primary human hepatocytes and HepG2 cells downregulates hepcidin mRNA.18,24 As iron absorption is enhanced in HH despite iron overload,1–3 and constitutive hepcidin expression prevented iron overload in a mouse model of haemochromatosis,25 we assume that hepcidin regulation is disrupted in HH patients. Depressed concentrations of hepcidin are obviously unable to inhibit sufficiently elevated intestinal iron absorption. Moreover, based on the findings that liver hepcidin expression is significantly decreased in the hfe knockout mouse11,22 and in HH patients,23 lack of pro-hepcidin upregulation in HH despite iron overload indicates that HFE may be involved in regulation of serum hepcidin levels.
Although previous studies have demonstrated that urinary hepcidin excretion correlates well with serum ferritin concentrations,24 in the present study no correlation was found between circulating pro-hepcidin and serum iron or ferritin levels in HH or dialysis patients. Likewise, no correlation was detected between pro-hepcidin and transferrin saturation, which is thought to regulate expression of liver hepcidin,18 although our HH patients were not affected by anaemia, hypoxia, or inflammation (hepcidin influencing factors). Therefore, we suggest that regulation of pro-hepcidin levels in serum by iron stores involves complex indirect effects.24
As hepcidin has also been isolated from urine, we were interested in hepcidin regulation in renal insufficient patients. In contrast with HH patients and healthy subjects, concentrations of immunoreactive pro-hepcidin in the serum of patients with CRI were significantly increased from 106.2 ng/ml in healthy subjects to 148.1 ng/ml. Enhanced levels of pro-hepcidin in dialysis patients suggests that the kidneys may be involved in metabolism and/or elimination of the circulating peptide. However, it is presently unclear if urinary hepcidin is filtered from blood or originates from the kidney. Based on our current studies, we cannot exclude the fact that hepcidin is released at least partly from the kidneys as it was also found in renal tubular cells.18
Interestingly, in a recent study on regulation of the hepcidin gene, the kidney hormone EPO was shown to downregulate liver hepcidin gene expression.26 Accordingly, EPO could act as an erythropoiesis stimulatory factor on the one hand and as a hepcidin inhibitory hormone on the other. Thus another explanation for enhanced hepcidin concentrations in dialysis patients could be the relative deficiency of EPO which is encountered regularly in terminal renal insufficiency.27,28 However, enhanced levels of pro-hepcidin were measured in our patients with CRI although they were treated with the hepcidin inhibitory hormone EPO, supporting the renal filtration of hepcidin.
In a recent study, hepcidin was discussed as a mediator of anaemia of inflammation as it was markedly induced by inflammation.24 In another study, inappropriate high expression of hepcidin was associated with refractory anaemia in patients with hepatic adenomas,29 suggesting that hepcidin may play a major causative role in the anaemia of chronic disease. In the present study, we determined pro-hepcidin serum levels in dialysis patients with RA, a well recognised complication of progressive renal failure which is characterised by normochromic normocytic erythrocytes. In comparison with healthy subjects, immunoreactive pro-hepcidin concentrations were not significantly higher in patients with RA (mean 115.0 ng/ml). Despite terminal renal insufficiency in these patients leading to accumulation of the peptide hormone, pro-hepcidin levels were significantly lower than in dialysis patients without anaemia (mean 148.1 ng/ml). From these findings we conclude that hepcidin regulation in RA is different from that in anaemia of inflammation or of hepatic adenomas. Downregulation of pro-hepcidin in RA may reflect reactive physiological modulation of the peptide to enhance intestinal iron absorption and iron release from reticuloendothelial macrophages. Reduced hepcidin levels in RA are in accordance with the situation in experimentally induced anaemia where the negative effect of anaemia on hepcidin gene expression was significantly dominant over the positive effect of iron.30 Indeed, in addition to impaired erythropoiesis as a result of inappropriate deficient production of EPO in CRI,27,28 renal anaemia is caused by blood loss during dialysis, occult bleeding in the gastrointestinal tract, diminished erythrocyte half life, haemolysis, and uraemic thrombocytopenia.27 EPO therapy in patients with RA could be related to hepcidin suppression but our findings in the group of patients with CRI without anaemia showed that pro-hepcidin was increased despite EPO therapy. Thus we assume that hepcidin is decreased in RA because of blood loss, which may be a reason for hepcidin downregulation.30
In summary, at present the molecular nature of the signals that programme enterocytes of the proximal intestine to adjust iron absorption remains to be identified. In this context, the recently detected peptide hormone, hepcidin, could act as a central player in the regulatory pathway from body iron stores to iron absorptive cells.8,31 However, to understand the role of hepcidin, knowledge about cellular origin is necessary. In this respect, we identified hepcidin immunoreactivity in human and guinea pig liver where it was localised to the basolateral membrane domain of hepatocytes.
We developed an ELISA to measure pro-hepcidin levels in human serum. This assay is non-invasive and easy to perform, and thus appropriate for routine work. The success of the pro-hepcidin assay is due to its precision, sensitivity, reproducibility, and exact determination of hepcidin-(28–47) in human serum samples. Application of the present ELISA allows, for the first time, detection and determination of pro-hepcidin in patients suffering from several disorders of iron metabolism. Further studies are required to identify the exact molecular mechanism of pro-hepcidin in various iron states. Nevertheless, we believe that hepcidin agonists and antagonists could provide potential drugs in the prevention and treatment of iron disorders.
Acknowledgments
We thank Dr M Franke for providing blood from patients with chronic renal insufficiency. The technical assistance of K Bents and A Fiedler is greatly acknowledged. This study was supported by grant STR 216/10-1 of the Deutsche Forschungsgemeinschaft, by the Dietmar-Hopp-Foundation, and by the Forschungsförderungs-Programm of the University Hospital Heidelberg (8/2001, 76/2002, 58 and 79/2003).
REFERENCES
Linked Articles
- Leading article
- Digest
- Commentary