Article Text

Abstract
Background/Aims Single-point mutations in the cationic trypsinogen gene have been reported in hereditary pancreatitis kindreds in the white population. The aim of the present study was to investigate whether similar gene mutations are present in Japanese hereditary pancreatitis kindreds.
Methods All five exons of the cationic trypsinogen gene were amplified by polymerase chain reaction and sequenced in six Japanese families with hereditary pancreatitis.
Results Two types of single-point mutation in the cationic trypsinogen gene, which were identical with those reported in white families with hereditary pancreatitis, were observed in separate Japanese families with hereditary pancreatitis: 21Asn (AAC) to Ile (ATC) (N21I) in exon 2 and 117Arg (CGC) to His (CAC) (R117H) in exon 3. Pancreatitis occurred at more advanced ages in patients with the N21I mutation than in those with the R117H mutation. Besides normal polymorphisms in exons 4 and 5, no mutation was found in patients in the remaining four families with hereditary pancreatitis, 21 patients with sporadic chronic pancreatitis, or five normal subjects.
Conclusions These results show heterogeneity, but no racial specificity, in the cationic trypsinogen gene mutations in hereditary pancreatitis kindreds. A distinctive clinical feature for each of the mutation types is suggested: adult onset for the N21I mutation and childhood onset for the R117H mutation.
- hereditary pancreatitis
- pancreatitis
- trypsinogen
- gene mutation
- Japanese
Abbreviations
- CT
- cationic trypsinogen
- HP
- hereditary pancreatitis
Statistics from Altmetric.com
Hereditary pancreatitis (HP) is an autosomal dominant disorder with 80% penetrance. Since the first description by Comfort and Steinberg in 19521 of a kindred group affected by HP, over 100 affected families from around the world have been described2 ,3; this has been reviewed by Le Bodicet al.4 The disease is characterised by recurrent episodes of pancreatitis often beginning in childhood, equal gender distribution, a family history of at least two other affected members, frequent presence of calcified stones in the pancreatic duct, and absence of known precipitating factors such as alcohol or gallstones.5 Recently, an increased cumulative risk of pancreatic cancer in elderly patients with HP, especially in individuals with a paternal inheritance pattern, was reported.6
In 1996, based on the results obtained from a genome-wide linkage analysis with highly informative microsatellite markers on a family extensively affected with HP, three independent research groups reported a tight linkage between the clinical phenotype of HP and chromosome 7q.7-9 Further, multipoint linkage analysis indicated that the HP gene is probably located on chromosome 7q35.8 ,9 At the same time, at least eight trypsinogen genes, including cationic and anionic trypsinogen genes and their pseudogenes, were found to be located within the gene cluster of the V and D-C segments of the T-cell receptor β chain on 7q35.10 Mutational screening analyses for each of the exons from the cationic and anionic trypsinogen genes were employed in multiple affected and unaffected members of a white HP family. A single G to A transition in exon 3 of cationic trypsinogen (CT) was identified in all affected members and obligate carriers in one HP kindred.11 It was predicted that this mutation would result in an Arg (CGC) to His (CAC) substitution at amino acid residue 117 of CT (using the chymotrypsin numbering system). An identical mutation has been found in five white HP families.11Another point mutation (A to T) in exon 2 of the CT gene was identified in separate white HP families, and it was predicted that this would result in an Asn (AAC) to Ile (ATC) substitution at amino acid residue 21 of CT.12
These findings prompted us to investigate whether Japanese HP kindreds of genetically different origin from white HP kindreds have the same mutations in the CT gene. We performed mutational screening analyses for all five exons of the CT gene in affected and unaffected members of six Japanese families with HP and also in patients with sporadic chronic pancreatitis. Mutations identical with those previously reported in white HP kindreds11 ,12 were found in exon 2 and 3 of the CT gene in separate Japanese HP families, indicating no racial specificity in CT gene mutations in patients with HP. Along with these genetic findings, clinical features relevant to the gene mutation types are also discussed in the present study.
Materials and methods
SUBJECTS
An HP family was defined by its meeting of the following criteria: at least three affected members, relatively early onset of pancreatitis, and the absence of other known precipitating factors of pancreatitis.5 All HP patients fulfilled the diagnostic criteria for chronic pancreatitis of the Japan Pancreas Society13 when enrolled in the present study. Onset of the disease was estimated on the basis of the patient’s statement about the clinical symptoms (upper abdominal pain and/or back pain). From the HP families nominated in the previous survey in 1995 by the research group for studying intractable pancreatic disease supported by the Ministry of Health and Welfare of Japan (published in Japanese in an annual report from the Japanese government, 1996), six HP families independently distributed all over Japan were selected for this study. All the selected families are of Japanese origin; other racial or ethnic families were not included. Peripheral blood samples were obtained from one or more affected members and, in one family, also from unaffected members (10 affected and two unaffected members in total). Twenty one patients with sporadic chronic pancreatitis (19 patients with idiopathic chronic pancreatitis and two with alcoholic chronic pancreatitis) who visited the hospital of Kochi Medical School during the period 1996–1997 and five normal subjects were studied. Definitive diagnosis of chronic pancreatitis was achieved on the basis of the criteria of the Japan Pancreas Society.13 All individuals studied gave informed consent according to the ethical guidelines of the Declaration of Helsinki.14
AMPLIFICATION OF EXONS IN THE CT GENE BY POLYMERASE CHAIN REACTION (PCR)
DNA was extracted from peripheral blood leucocytes with a QIAamp blood kit (Qiagen, Hiden, Germany). All five exons of the CT gene were individually amplified by PCR. Primer sets for PCR specific for the CT gene were designed and synthesised to include both sides of the intronic flanking sequence of each exon for analysing splicing mutations. Because of the extremely high homology of the intronic sequence of the CT gene with that of pseudogenes, basically a nested PCR strategy and/or a cycle sequence with an inner primer were employed, as shown in table 1. Tables 1 and 2 show primer sequences and suitable annealing temperatures. PCR amplification was performed using a commercial kit (TaKaRa, Kyoto, Japan). The PCR amplification programme was as follows: a hot start (95°C, three minutes) and an additional 40 cycles (at 94°C for 30 seconds for denaturing, at the specified temperature for each primer set (see table 2) for 30 seconds for annealing, and at 72°C for two minutes for extension), followed by an incubation at 72°C for 10 minutes.
Primer sets for PCR and cycle sequencing
Primer sequences
DNA SEQUENCING OF PCR PRODUCTS
PCR-amplified products were electrophoretically separated on a 2% agarose gel. Bands of appropriate size were cut from the gel, purified with the GENE Clean III Kit (Bio 101, Vista, California, USA), and sequenced by using the ABI PRISM Dye Termination Cycle Sequencing Kit (Perkin-Elmer, Foster City, California, USA) and an ABI 370A DNA Sequencer (ABI, Foster City, California, USA). The sequences obtained for each exon of the CT gene were double checked by sequencing with forward and reverse flanking primers.
COMPUTER ANALYSIS FOR THE MOLECULAR NATURE OF WILD TYPE AND MUTANT CT
Using the predicted amino acid sequences of wild type and mutant CT, cleavage sites for proteolytic enzymes and chemicals were analysed and Chou-Fasman and Robson-Garnier secondary structure predictions were made using a computer analysis program (MacVector 3.5, International Biotechnologies Inc, Rochester, New York, USA).
Results
SEQUENCE ANALYSIS OF CT
All five exons of the CT gene along with their flanking introns were amplified by PCR and directly sequenced in affected and unaffected members of six HP families. In two of the six families, different types of single-point mutation were identified. In one of these families (designated number 1000, see the pedigree in fig 1A), a heterozygous mutation, G to A, in exon 3 was found in three affected members but not in two unaffected members. It was predicted that this point mutation would result in an Arg (CGC) to His (CAC) substitution at amino acid residue 117 of CT (using the chymotrypsin numbering system) (fig 2). In the other family (designated number 6000, see the pedigree in fig 1B), a heterozygous point mutation, A to T, in exon 2 was identified in two affected members, and it was predicted that the mutation would result in an Asn (AAC) to Ile (ATC) substitution at amino acid residue 21 (fig3). These point mutations were identical with the mutations previously reported in white HP kindreds11 ,12 and were not observed in patients in the remaining four HP families, 21 patients with sporadic chronic pancreatitis, or five normal subjects.
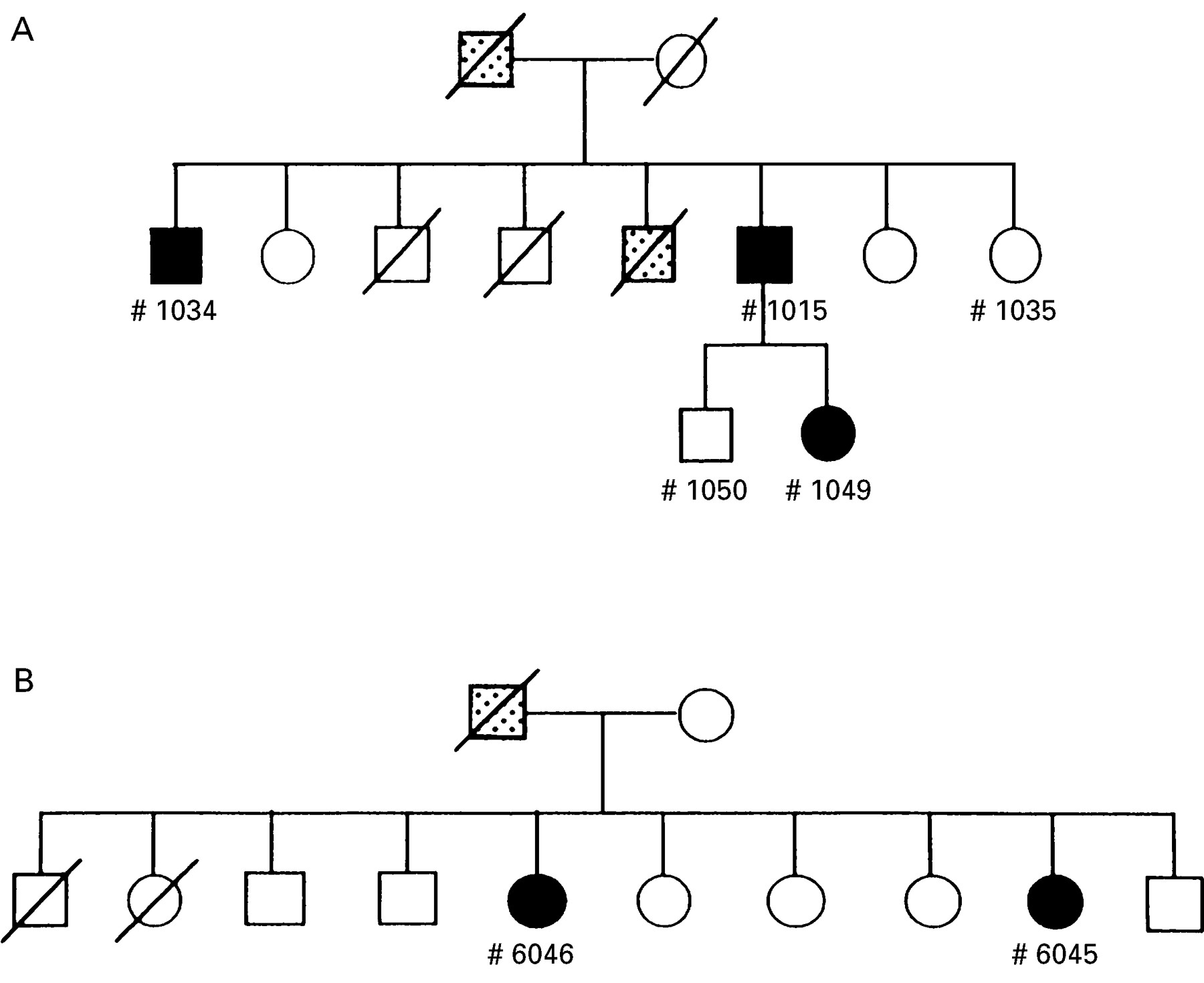
Pedigrees of patients with hereditary pancreatitis (HP) with the mutation in the cationic trypsinogen gene. In one HP family (A; family number 1000), the R117H mutation in exon 3 was found in three affected members (1015, 1034, and 1049; see fig 2) but not in two unaffected members (1035 and 1050). In another HP family (B; family number 6000), the N21I mutation in exon 2 was identified in two affected members (6046 and 6045; see fig 3). A closed square or circle indicates a patient with pancreatitis, and a stippled symbol indicates family members suspected of having pancreatitis based on available clinical records.

DNA sequencing electropherograms of the human cationic trypsinogen gene exon 3 in the region of the mutation identified in patients with hereditary pancreatitis (HP) (A, forward sequencing; B, reverse sequencing). Heterozygosity at the second nucleotide in the frame (G/A) was shown in three affected members but not in two unaffected members in HP family 1000 (see the pedigree in fig 1A). This point mutation was predicted to result in an Arg (CGC) to His (CAC) substitution.

DNA sequencing electropherograms of the human cationic trypsinogen gene exon 2 in the region of the mutation identified in patients with hereditary pancreatitis (HP) (A, forward sequencing; B, reverse sequencing). Heterozygosity at the second nucleotide in the frame (A/T) was shown in two affected members in HP family 6000 (see the pedigree in fig 1B). This point mutation was predicted to result in an Asn (AAC) to Ile (ATC) substitution.
It should be noted that single C to T transitions in exons 4 and 5 without amino acid substitution (162Asp (GAC-GAT) and246Asn (AAC-AAT)) were identified in patients of all six HP families. However, these transition types were also observed in unaffected members of an HP family, patients with sporadic chronic pancreatitis, and normal subjects.
CLINICAL FEATURES IN HP PATIENTS WITH CT GENE MUTATION
Clinical features were analysed in three patients with the R117H mutation (family number 1000) and two patients with the N21I mutation (family number 6000) (table 3). Unfortunately, details of clinical records of other patients in both HP families were not available, since they had died before the study. In patients in the former family, age of onset of pancreatitis was five, six, and 16 (average age nine). In contrast, the two patients in the second family had a relatively high age of onset of pancreatitis, 34 and 28. Available clinical records indicated that no patient died from pancreatic cancer in either family.
Clinical features of patients with hereditary pancreatitis in two families with the gene mutation in cationic trypsinogen
COMPARISON OF THE MOLECULAR NATURE OF WILD TYPE AND MUTANT CT
Using the predicted amino acid sequences of wild type and mutant (N21I) CT, cleavage sites for proteolytic enzymes and chemicals and the secondary structure were analysed. Computer analysis showed that the N21I substitution did not remove any of the cleavage sites (data not shown). However, secondary structure predictions showed that the N21I substitution altered the native turn structure of the flanking region of wild type CT molecules to a sheet structure on mutant CT (fig 4). It is noteworthy that 21Ile is found in the corresponding amino acid sequence of anionic trypsinogen, which is another of the trypsinogen isoforms,15 meaning that the 39 amino acid flanking sequence containing 21Ile (residues 15–63) on the mutated CT (see fig 4B) is identical with that of anionic trypsinogen.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Chou-Fasman and Robson-Garnier secondary structure predictions on the predicted amino acid sequences of wild type and mutant (N21I) cationic trypsinogen (CT). Computer analysis showed that the N21I substitution (arrowheads) alters the native turn structure of the flanking region of wild type CT (A) to sheet structure on mutant CT (B). It is noteworthy that 21Ile is found in the corresponding amino acid sequence of anionic trypsinogen, which is another of the trypsinogen isoforms, meaning that the amino acid sequence presented in B (39 residues long) is identical with that of anionic trypsinogen. CF, Chou-Fasman prediction; RG, Robson-Garnier prediction; CfRg, Chou-Fasman and Robson-Garnier combined prediction; Helix or Hlx, helix structure; Sheet or Sht, sheet structure; Turns or Trn, turn structure.
Discussion
Two types of single-point mutation in the CT gene were identified in two separate Japanese families with HP. These mutation types were identical with those previously reported in several white HP families.11 ,12 One of them, the mutation117Arg (CGC) to His (CAC) in exon 3, was reported to be found in four HP families in the United States and one family in Italy.11 Another research group in Japan also preliminarily described the same mutation in three Japanese HP families.16 Another mutation, 21Asn (AAC) to Ile (ATC) in exon 2, was found in two HP families in the United States12 and preliminarily reported in one family in Germany.17 The fact that identical mutations were observed in mongolian and white HP kindreds indicates heterogeneity, but no racial specificity, in CT gene mutations in patients with HP. It may be that these types of mutation are present world wide.
In the present study, a single C to T transition without amino acid substitution was identified in exons 4 (162Asp) and 5 (246Asn) in some patients with HP or sporadic chronic pancreatitis and also in some normal subjects. In a comparison of the cDNA sequence of CT with its genomic DNA sequence, which has been independently reported,10 ,15 three sites of a single base transition with no amino acid substitution were identified, one in exon 4 and the other two in exon 5 (data not shown). Two of the transitions were identical with the transitions found in the present study. These single base substitutions in exons 4 and 5 were reported in a preliminary mutational analysis of the CT gene in HP patients in Germany.17 Gorry et al 12 also reported a possible normal polymorphism in exon 4 of the CT gene. These findings suggest that the single C to T transitions in exons 4 and 5 are normal polymorphisms.
On the basis of x ray crystal structure analysis, molecular modelling, and protein digest data, Whitcombet al 11 indicated that the117Arg residue is a trypsin-sensitive site and provided a diagram of a model of the trypsin self-destruct mechanism designed to prevent pancreatic autodigestion. Certain enzymes, including mesotrypsin, enzyme Y, and trypsin itself, are postulated to be part of a feedback mechanism for inactivating wild type trypsin by cleaving its molecule. If the 117Arg cleavage site is replaced by His, prematurely activated trypsin would not be destroyed, leading to autodigestion of the pancreas. Although this hypothesis has not been proved, it is a likely explanation of why the R117H mutation in the CT gene results in pancreatitis as a hereditarily determined clinical phenotype. However, this hypothesis cannot be applied to the N21I mutation, since this type of amino acid substitution did not result in the disappearance of the cleavage site for proteolytic enzymes and chemicals on the CT molecule.
Another possible explanation is that the amino acid substitution caused by the mutation alters the conformational structure of CT which may reduce its ability to bind to pancreatic secretory trypsin inhibitor. Even though the analysis is based on a linear sequence, it was predicted that the N21I substitution alters the native turn structure of the flanking region on the CT molecule to sheet structure (fig 4). Gorry et al 12 also suggested that some kind of conformational alteration may impair trypsin inactivation by altering the accessibility of 117Arg to trypsin or protecting the adjacent C22–C157 disulphide bond to prolong survival of trypsin after limited hydrolysis. However, the 39 amino acid sequence on the mutant CT containing the N21I substitution is identical with the corresponding sequence of anionic trypsinogen, which is another isoform of trypsinogen. Thus it is questionable whether this type of mutation on CT would dramatically alter its molecular nature such as blocking a cleavage site or reducing its ability to bind to trypsin inhibitor. The hypothesis that the CT gene mutations produce pancreatitis is still open to question. Further studies includingx ray crystal structure analysis of the mutated CT molecule and CT mutation-transgenic animals will be required.
Comparison of the clinical features of the two families with the different types of CT gene mutation suggests a notable difference: age of disease onset. HP patients with the N21I mutation had a relatively higher age of onset than those with the R117H mutation. These findings are consistent with the previous report by Gorry et al 12 that a high percentage of patients with a higher age of disease onset was observed in an HP family with the N21I mutation compared with an HP family with the R117H mutation. Because of the small number of patients with each mutation type in the present study, however, final conclusions should not be drawn.
The present study showed heterogeneity, but no racial specificity, of CT gene mutation in HP. Identification of CT gene mutation will enable us to study the pathophysiology of HP. Furthermore, clarification of the pathophysiology of HP may lead to a breakthrough in our understanding of sporadic cases of chronic and acute pancreatitis. Thus it is important to know the clinical features of pancreatitis in relation to the CT gene mutation type, including any increased risk of pancreatic cancer. Together with a worldwide genetic survey of HP kindreds, detailed investigations of clinical features involving large numbers of patients and kindreds are urgently required.
Acknowledgments
This work was supported by a grant from the Japanese Ministry of Health and Welfare for the study of intractable pancreatic disease.
Abbreviations
- CT
- cationic trypsinogen
- HP
- hereditary pancreatitis
References
Linked Articles
- Commentary