Article Text

Abstract
AIM To contribute to the establishment of a rational clinical, neuroradiological, and molecular approach to neurogenic muscle weakness, ataxia, and retinitis pigmentosa (NARP) and maternally inherited Leigh’s syndrome (MILS).
METHODS AND RESULTS The T8993G mutation in the mitochondrial genome was found in several maternal members of six pedigrees, whose clinical status ranged from no symptoms to severe infantile subacute necrotising encephalomyelopathy (Leigh’s disease). In one case a MELAS-like syndrome was documented both clinically and neuroradiologically. Relevant genetic features of the series were anticipation of symptoms through subsequent generations, and the presence of several cases in whom the mutation apparently occurred recently or was new. A uniform distribution of the mutation in many tissues was shown in one patient subjected to necropsy. In general, a good correlation was found between clinical severity and mutation heteroplasmy in readily accessible tissues, such as lymphocytes or fibroblasts. By contrast, a consistent reduction of the mitochondrial ATPase activity, to about half of the normal values, was found in most of the clinically affected cases, irrespective of the amount of mutant mitochondrial DNA.
CONCLUSIONS Although the measurement of ATP hydrolysis in cultured fibroblasts was a reliable, and sometimes instrumental, means to identify T8993G positive patients, the relation between the mutation and the oxidative phosphorylation defect is probably very complex, and its understanding requires more complex biochemical analysis.
- Leigh’s syndrome
- NARP
- ATPase
Statistics from Altmetric.com
Combined peripheral neuropathy, ataxia, retinitis pigmentosa, seizures, and dementia are the clinical features of NARP (neurogenic muscle weakness, ataxia,and retinitis pigmentosa), a maternally inherited, adult onset disorder associated with a heteroplasmic T to G transversion at mtDNA nucleotide position (np) 8993.1 The T8993G change converts a conserved leucine into arginine (L155R) in subunit 6 of mitochondrial F0F1-ATP synthase. However, the T8993G mutation has also been found in several cases of subacute necrotising encephalomyelopathy (Leigh’s syndrome) with normal cytochrome c oxidase and pyruvate dehydrogenase activity (MILS, maternally inherited Leigh’s syndrome).2-5 Both syndromes can be present in the same family.6 7 A second mutation—T8993C transition—causing a proline for leucine replacement (L155P), was later reported in one NARP/MILS family,8 and confirmed in one patient with Leigh’s syndrome.9 Both amino acid changes occurred in a charged region associated with the proton channel of ATP synthase,10 11 possibly interfering with the utilisation of the transmembrane electrochemical gradient to produce ATP.2
Several inherited neurodegenerative disorders share some, or most, of the clinical hallmarks of NARP. For instance, hearing loss, ataxia, and visual impairment due to retinitis pigmentosa are features common to NARP, Refsum’s disease, and Usher’s syndrome. The similarity between NARP and non-mitochondrial neurodegenerative disorders is made even stronger by the lack of ragged red fibres , or other histological “mitochondrial” hallmarks in muscle biopsies of patients with NARP.1 Lactic acidosis may be absent as well. Muscle weakness is often mild and masked by the cerebellar symptoms, and the biochemical defect associated with NARP may go undetected because ATPase activity is seldom measured in routine screening of respiratory chain defects.
As a contribution to the establishment of a rational clinical, neuroradiological, and molecular approach to these disorders, we report here six new T8993G-positive maternal lineage, associated with NARP, MILS, or both.
Case reports
Figure 1 shows the pedigrees of our patients. Table 1 summarises the clinical and genetic features.

Pedigree trees. Percentages indicate the proportion of mutant mtDNA in blood lymphocytes of all members with the exception of patients III/1, pedigree B and II/1, pedigree E for whom values from muscle mtDNA analysis are reported. Black solid symbols=early onset Leigh’s syndrome; three quarter black symbols=late infancy intermediate clinical syndrome; half black symbols= NARP syndrome; one quarter black symbol=a woman with a “minimal syndrome” (see text); open symbols=unaffected, reportedly unaffected, or clinically unknown members; small black solid circles=miscarriages; open triangle in pedigree C=three deceased members for whom no other information was available.
Clinical and molecular genetic data
PEDIGREE A
A boy (IV/2), born after a full term pregnancy from healthy, unrelated parents, presented in the first months of life with a severe psychomotor delay, swallowing and respiratory difficulties, and incoordination of the ocular movements. Neurological examination showed an infant with no visual contact, little spontaneous movement, severe generalised hypotonia, and brisk tendon reflexes. Fundus examination was normal. He had metabolic acidosis, due to high concentrations of lactate in blood (3.3 mM) and CSF (5.6 mM; normal values in both compartments <1.8 mM). Brain CT showed bilateral, symmetric areas of low density in the basal ganglia. An EEG showed multifocal, asynchronous spike and wave discharges. A muscle biopsy showed mild “myopathic” changes, with no ragged red fibres or other signs of mitochondrial involvement. The child died of central respiratory arrest at 7 months of age, after a rapidly deteriorated course. No case of neuromuscular disorder was reported in the family. The mother of the proband is now 36 years old and healthy.
A necropsy examination of the brain showed severe degeneration of the caudate nucleus, putamen, globus pallidus, and substantia nigra. These regions were characterised by pronounced spongiosis, astrocytosis, and neuronal loss, associated with vascular proliferation. Scattered areas of pronounced vacuolisation were also present.
PEDIGREE B
A girl (III/1), born after a full term pregnancy from unrelated parents, had generalised hypotonia and severe psychomotor delay from the first months of life. At 6 months of age, during a febrile illness, she developed metabolic acidosis, respiratory difficulty, and generalised seizures. Neurological examination showed an infant with no visual contact, severe hypotonia, reduced spontaneous activity, and dystonic postures of the limbs. Lactate concentrations were raised in blood (4.3 mM). Brain CT showed bilateral, symmetric low density areas in the basal ganglia, and cortical atrophy. The visual and brainstem auditory evoked potentials (VEPs and BAEPs) were abnormal. No case of neuromuscular disorder was reported in the family. However, the neurological examination of the proband’s mother, now 34 years old, showed moderate ophthalmoparesis, mild bilateral pes cavus, and slightly increased latency of VEPs. We considered her as affected, with “minimal signs”.
PEDIGREE C
The index case was a girl (III/1), born after normal pregnancy and delivery. In the first months of life she showed generalised hypotonia with brisk tendon reflexes, severe psychomotor delay, and progressive swallowing difficulty. At 1 year of age she had microcephaly and developed epileptic seizures. A neurological examination at the age of 4 showed severe hypotonia, distal muscular atrophy, no spontaneous motor activity, and no visual contact. The concentrations of lactate were increased in both blood (4.9 mM) and CSF (3.1 mM). The fundus examination and VEPs were normal. The BAEPs disclosed bilateral abnormalities of central conduction. An MRI showed cerebellar and cerebral atrophy and symmetric hyperintense signal abnormalities of basal ganglia and brainstem on T2 weighted images (fig 2 A and B). The morphological examination of the muscle biopsy failed to identify myopathic abnormalities. The patient was the first offspring of a 24 year old, moderately affected woman, belonging to a family composed of four siblings (II/1–4, fig 1). The siblings were all variably affected by juvenile onset ataxia, weakness, hearing loss, retinitis pigmentosa, and mental deterioration. As they were born from a healthy mother, they were diagnosed as having an autosomal recessive disorder, most likely Usher’s syndrome. A brain MRI examination, performed in patients II/3 and II/4, showed moderate, diffuse cerebral and cerebellar atrophy. The atrophy was more pronounced in the severely affected patient II/3, who also showed focal symmetric lesions of the basal ganglia. The MRI of their clinically healthy mother (I/1, fig 1) was normal. Nerve conduction velocities were normal whereas sensory action potentials (SAPs) were of low amplitude indicating an axonal sensory neuropathy. VEPs and BAEPs were abnormal in the two examined affected sisters, whereas they were normal in their mother.

MRI examination of patient III/1, pedigree C. (A) a T1 weighted midsagittal section shows diffuse atrophy particularly involving the inferior vermis of the cerebellum. (B) A T2 weighted axial section shows symmetric hyperintense signal involving the head of the caudate nucleus, the putamen, and part of the pallidum. The medial part of the thalamus is also involved (arrows).
PEDIGREE D
The proband (III/1) was a 9 year old boy, born at term after a normal pregnancy from unrelated parents. The family history was unremarkable. From the age of 5 years, the patient had a progressive neurological syndrome, characterised by mild psychomotor delay, unsteadiness of gait, and slurred speech. At the age of 7 years, during an acute febrile illness, he presented with vomiting, progressive generalised weakness, and severe ataxia. At 9 years he was severely dysmetric, ataxic, and dysarthric. He had dystonic movements and mild mental deterioration. Fundus examination was normal. However, electroretinography (ERG) showed potentials of reduced amplitude. VEPs showed increased latency of P100, and BAEPs were normal. The EMG and nerve conduction velocities were also normal. Plasma lactate and pyruvate were normal but plasma alanine was abnormally raised (0.472 mM; normal values<0.360). Brain MRI showed the presence of symmetric, hyperintense alterations in the putamen on T2 weighted images, and cerebellar and cortical atrophy. A muscle biopsy showed mild myopathic changes, but no mitochondrial abnormalities. The mother of the proband is now 32 years old and clinically healthy.
PEDIGREE E
The proband (II/1) was a boy born after normal pregnancy and delivery. He showed normal psychomotor development until 4 years of age when, during a febrile illness, he presented with an acute ataxic syndrome associated with visual impairment and generalised epileptic seizures. At 8 years of age, fundus examination showed retinitis pigmentosa. An ERG was abnormal. Brain MRI disclosed bilateral abnormal signal intensity in the basal ganglia and cerebellar atrophy.
At the age of 12, during an episode of febrile illness, he had a rapid worsening of neurological symptoms, including hypotonia and muscle weakness, both more severe on the left side, a bilateral Babinski’s sign, and generalised seizures. An MRI showed, in addition to the cerebellar atrophy and abnormalities of the basal ganglia, a wide area of abnormal signal intensity, involving the right occipital and parietal cortex and subcortical white matter; an irregular area of abnormal signal intensity was also present in the right thalamus. The entire parieto-occipital region was swollen, compressing the right lateral ventricle (figure 3 A and B).
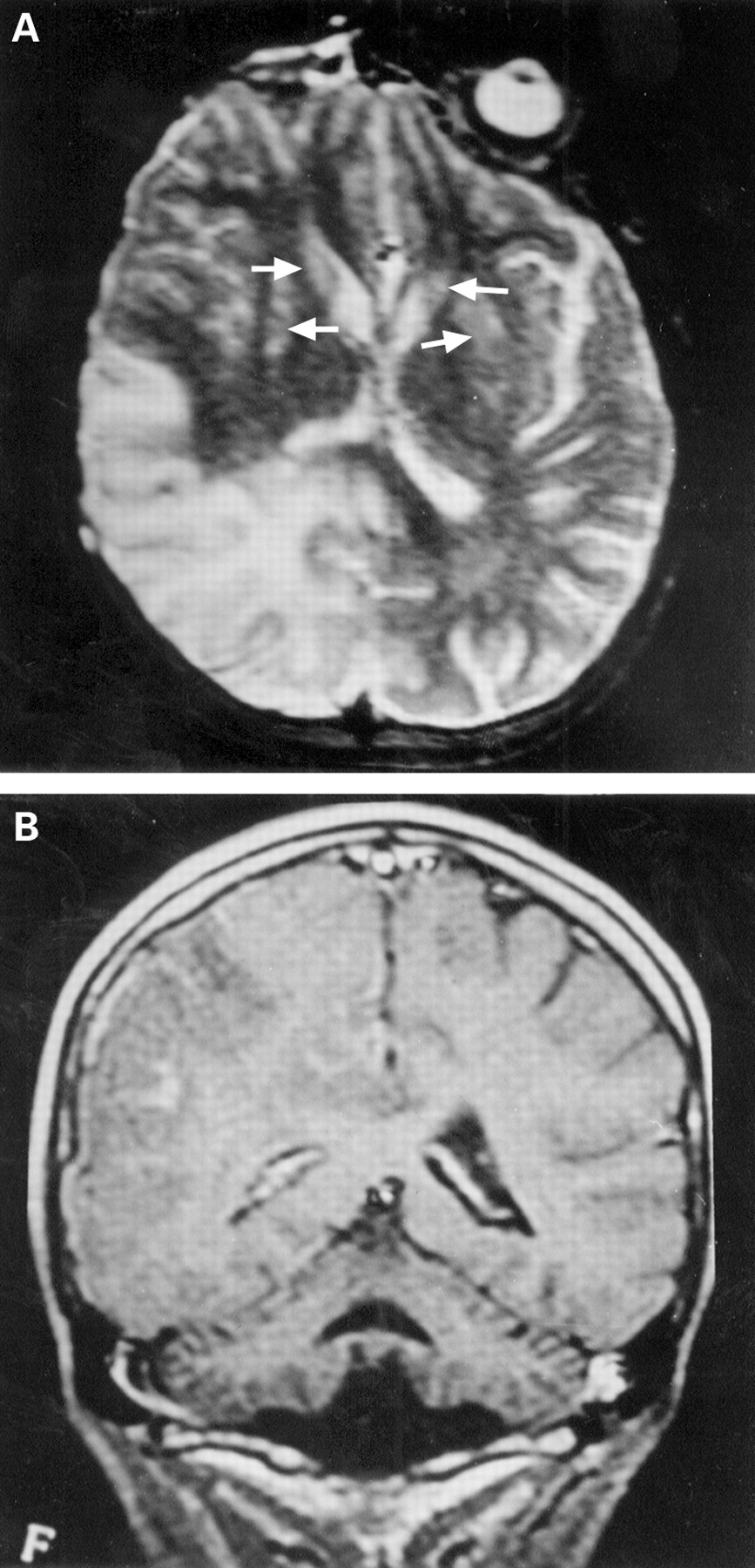
Brain MRI of patient II/1, pedigree E. (A) a T2 weighted axial section shows hyperintense signal involving the posterior part of the right hemisphere and anterior displacement of the atrium of the right lateral ventricle. Signal abnormalities are also present in the neostriatum bilaterally (arrows). (B) a T1 weighted coronal section after administration of contrast medium (Gd-DTPA) of the same patient shows atrophy of the cerebellum and the large lesion of the right hemisphere with scattered areas of superficial and subcortical enhancement.
No signs of cardiomyopathy, abnormalities of cardiac rhythm, or other cardiogenic causes of brain thromboembolism were found by clinical examination or ECG. A diagnosis of MELAS was considered and, despite normal concentrations of lactate and pyruvate in plasma and CSF, a muscle biopsy was performed. Neither ragged red fibres nor succinate dehydrogenase hyperintense vessels were found by light microscopy examination. Analysis of mtDNA for MELAS and MERRF mutations were negative.
Neurological conditions remained stable and CT at the age of 13 showed the presence of multiple, bilateral lesions in the brain cortex, and diffuse atrophy of the brain, cerebellum, and brainstem. The BAEPs and VEPs were abnormal, and the ERG response was absent. Sensory nerve conduction velocities were slightly reduced with a low amplitude SAP.
The mother of the proband, now 29 years old, as well as his 13 and 9 year old sisters, are clinically healthy.
PEDIGREE F
The proband (IV/5) was born at term after a normal pregnancy and delivery. At 7 months of age mild, progressive psychomotor delay was noticed. He was able to walk without support at 30 months of age. Verbal skills and intellectual performances were very delayed. At 5 years of age an MRI examination of the brain showed mild atrophy of the cerebellar vermis, with no abnormal signal intensities. Progressive visual loss had been noticed since the age of 7 years. At the age of 9, neurological evaluation showed cerebellar ataxia, bilateral pyramidal signs, severe visual loss, and mental retardation. Fundus examination disclosed retinitis pigmentosa. VEPs and ERG were abnormal. Nerve conduction velocities were low, indicating sensorimotor peripheral neuropathy.
Although the maternal lineage was negative for neuromuscular disorders, the proband’s mother (III/10, fig 1) developed rapidly progressive visual loss at the age of 32, associated with retinitis pigmentosa and later steadily worsening ataxia. Moreover, she had two miscarriages, before giving birth to a second child, now three years old and clinically healthy. Brain MRI of patient III/10 showed two areas of abnormal signals in the periventricular white matter, and oligoclonal bands were seen in the CSF.
Methods
DNA ANALYSIS
Total DNA was extracted from blood lymphocytes, cultured fibroblasts, or muscle tissue, according to standard methods. In one case, (proband IV/2, pedigree A) the DNAs from several tissues obtained at necropsy were examined for the T8993G mutation, including muscle, heart, liver, kidney, frontal cortex, basal ganglia, thalamus, cerebellum, and brainstem.
The presence of the NARP associated mtDNA mutations was investigated by restriction fragment length polymorphism (RFLP) analysis on mtDNA fragments amplified by the polymerase chain reaction (PCR). Oligonucleotides used for PCR priming were as follows: (A) np 8650–8675 (sense); (B) np 9155–9180 (antisense). The cycle conditions were as follows: denaturation for 60 seconds at 94°C; annealing for 60 seconds at 58°C; extension for 60 seconds at 72°C. The 530 bp DNA fragments were then exposed to the restriction endonucleases AvaI (specific to the T8993G transversion mutation1) or Hpa II (specific to the T8993C transition mutation8). In the presence of either mutation, the 530 bp PCR fragment was digested into two fragments of 343 and 187 bp, that could be separated from the undigested, wild type fragment by electrophoresis through a 2% agarose gel, and visualised with ethidium bromide under a UV transilluminator.
The amount of digested versus undigested DNA fragments was measured with a densitometer.
In a second set of experiments, Southern blot analysis of the AvaI digested fragments was carried out, using the same 530 bp PCR DNA fragment as a probe. The ECL chemiluminescence kit (Amersham) was used to label the probe with peroxidase, and to detect the fragments by autoradiography, according to the instructions of the manufacturer.
The presence of the T8993C mutation was confirmed by sequence analysis in two cases (IV/2, pedigree A; III/1, pedigree C).
BIOCHEMICAL ANALYSIS
Rotenone sensitive NADH coenzyme Q1 reductase (complex I), succinate dehydrogenase (part of complex II),12 cytochrome c oxidase (complex IV),13 antimycin sensitive ubiquinol cytochrome c reductase (complex III),14 oligomycin sensitive ATPase (complex V),15 and citrate synthase16 were measured in duplicate in muscle homogenates, as well as in cultured fibroblasts.
For biochemical assays, fibroblasts were treated with digitonin and subjected to three freezing and thawing cycles, as described elsewhere.17
The activities of the respiratory chain enzymes were normalised to the number of mitochondria by dividing each specific activity with that of citrate synthase.
Results
DNA ANALYSIS
All the proband infants showed the presence of a heteroplasmic T8993G transversion mutation in the mtDNA from blood lymphocytes, cultured fibroblasts, or muscle biopsy (table 1). The mutation was about 95% in the muscle DNA of the most severely affected patients (IV/2, pedigree A; III/1, pedigree B; III/1 pedigree C). The mutation was in slightly lesser amount (about 80%) in different tissues from the infants that showed a later onset and a slower, more progressive course of the disease (III/1, pedigree D; II/1, pedigree E; IV/5, pedigree F).
In the adult patients affected by NARP belonging to the sibship of pedigree C the percentage of mutation in blood lymphocytes ranged from 78% in a severely affected brother (II/2) to 58% in a mildly affected sister (II/4). The mutation was present at low percentage (19%) in the lymphocyte DNA of their healthy mother (I/1, maternal grandmother of proband III/1). Likewise, a low percentage of mutation (30%) was detected in blood lymphocyte mtDNA of the healthy mother of proband III/1, pedigree D. However, mutant mtDNA was 65% in lymphocytes of the very mildly affected mother of proband III/1, pedigree B and was not detected in lymphocytes of the maternal grandmother. Likewise, no mutation was present in lymphocyte mtDNA of the maternal grandmother of patient III/1, pedigree D. The mutation was virtually absent in the lymphocyte and fibroblast mtDNA of the healthy mother of proband IV/2, pedigree A, as well as in the mother and sisters of proband II/1, pedigree E. An extensive screening in lymphocyte DNA of numerous members of pedigree F failed to detect the mutation in any of the maternal relatives of the proband (IV/5, 85% mutation), with the exception of his moderately affected mother (III/10, 55% mutation) and clinically healthy younger sister (IV/6, 65% mutation). Absence of mutation in all samples that resulted negative by direct, ethidium bromide based RFLP detection was confirmed by Southern blot analysis using a chemiluminescence based detection kit that, according to the manufacturer, can show as little as 0.5 pg hybridising DNA (data not shown).
Finally, heteroplasmy varied very little in subjects in whom the T8993G mutation was investigated in several tissues. The most striking example was in the case of numerous necropsy samples (heart, liver, kidney, cerebellum, brain) of proband IV/2, pedigree A, that all showed the same amount of mutation ( 90%–95%).
BIOCHEMICAL RESULTS
Table 2 shows the activities of ATPase, measured as a reaction linked to pyruvate kinase and lactate dehydrogenase15 and normalised to that of citrate synthase of several samples from different members.
Complex V activity
A reduction of ATPase activity was found in cultured fibroblasts of all of the tested patients, ranging from 35% in patient IV/2, pedigree A, to 69% in patient III/1, pedigree C. However, the ATPase activity in muscle was within the normal range in several samples of the same patients that showed the defect in fibroblasts (III/1, pedigree B; III/1, pedigree D). Other respiratory chain activities were generally normal, with the exception of patient IV/2, pedigree A, whose muscle biopsy showed a slight reduction of cytochrome c oxidase/ citrate synthase (40, normal values 60–150) and succinate dehydrogenase/citrate synthase (8.0, normal values 11.5–19.3).
Discussion and conclusions
CLINICAL CONSIDERATIONS
Clinically, our T8993G positive patients can be divided into three different forms: early onset Leigh’s syndrome, a late infancy syndrome of intermediate severity, and adult onset NARP (table 1).
Three of our patients were affected by severe Leigh’s syndrome. The clinical picture was dominated by early onset, rapidly progressive failure of brainstem and cerebellar functions, leading to death or vegetative status, in a few months after birth. Retinitis pigmentosa was consistently absent.
The intermediate clinical presentation was characterised by pronounced clinical variability. Onset was in infancy, but usually later than in Leigh’s syndrome. As in the adult syndrome, the main symptoms were progressive visual impairment due to retinitis pigmentosa and cerebellar dysfunction. Additional features were present in individual cases. For instance, our patient II/1, pedigree E, presented an acute, stroke like episode, documented by MRI examination of the brain, suggesting a MELAS syndrome. However, the absence of the most common MELAS-associated mutations, and the discovery of a partial defect of mitochondrial ATPase activity in the patient’s muscle, led us to the correct diagnosis.
The affected siblings of pedigree C included three female patients with a full blown, adult onset NARP syndrome. They were born from a healthy, intelligent woman, suggesting an autosomal recessive trait, possibly Usher’s syndrome (McKusick n 276 900). This is characterised by vestibular ataxia, hearing loss, retinitis pigmentosa, and mild mental deterioration.18 However, the first child of a moderately affected female patient of pedigree C was a baby girl, patient III/1, presenting with Leigh’s syndrome. An isolated defect of oligomycin sensitive ATPase activity, that is carried out by complex V, led to the identification of the T8993G mutation in her muscle and fibroblasts. Lesser amounts of the same mutation were then found in blood lymphocytes of the baby’s mother, of the mother’s affected siblings, and, in even lower percentage, of the healthy grandmother. Thus a correct diagnosis could be given retrospectively to this family, because of the birth of a child with the most severe variant of the NARP phenotype.
The correction of the clinical diagnosis, from an allegedly autosomal recessive, to an experimentally proved maternally inherited aetiology, has important implications on the clinical management and genetic counselling in this family.
In one adult case the presence of oligoclonal bands in the CSF suggested a demyelinating disorder similar to the association that Harding et al 19 found in female patients between multiple sclerosis and a mtDNA mutation associated with Leber’s hereditary optic neurodegeneration. Whether the NARP mutation can contribute to produce the multiple sclerosis-like findings in our patient remains an interesting but as yet unproved hypothesis.
GENETIC CONSIDERATIONS
In two of our six families (pedigrees C and F), more than one generation was clinically affected. Onset of disease occurred earlier, and symptoms worsened from one generation to the next, in parallel with the increase of the T8993G mutation heteroplasmy (fig 1). Mutation heteroplasmy increased abruptly in two additional families (pedigrees B and D) from a clinically healthy (or very mildly affected) mother, to a severely affected child patient. Mutation was absent in the maternal grandmothers, indicating recently occurred mutational events. In a fourth pedigree (pedigree F) this phenomenon was virtually demonstrated by the absence of mutation in the maternal great grandmother, as well as in all of the tested maternal relatives of the proband’s mother. The mutation was absent in lymphocyte DNA of the mother and sisters of patient II/1, pedigree E, and in lymphocyte and fibroblast DNA of the mother and grandmother of patient IV/2, pedigree A. These results suggest either new events in the affected children, or the presence of a very skewed distribution of the mutation in the patients’ mothers, leading to germinal mosaicism. In summary, in four out of six pedigrees the proband child was the only clinically affected member; in five pedigrees mtDNA analysis suggested that the mutation raised de novo either in the proband or in the proband’s mother. From a practical point of view, these data indicate that an mtDNA aetiology can be suspected irrespective of the presence of obvious maternal transmission of the trait. A replicative advantage for mitochondria harbouring the T8993G mutation has been invoked to explain its rapid expansion along the generations.7 However, it is still possible that if the percentage of a mutation remains consistently below the phenotypic threshold, it may eventually disappear without ever being detected.
Interestingly, we detected similar amounts of T8993G mutation in several tissues taken at necropsy from a patient with Leigh’s disease, and little fluctuation of heteroplasmy was found in lymphocyte, fibroblast, and muscle mitochondria of several members. Similar results were reported by Ciafaloni et al 3 in a T8993G positive patient with MILS with >95% mutant mtDNA in fibroblasts, muscle, nerve, and brain. These data suggest that quantitative analysis of the T8993G mutation heteroplasmy can be performed reliably in readily accessible sources of DNA, such as peripheral blood or extrafetal tissues.20
GENOTYPE TO PHENOTYPE CORRELATIONS
As for other mtDNA mutations, the phenotypic threshold of the T8993G mutation is relatively high (>60%) suggesting a “recessive-like” effect. Despite extensive overlap among different groups of patients, the clinical status of our cases was clearly dependent on the presence and amount of mutation. As illustrated in fig4, the age of onset, reflecting clinical severity, and the percentage of heteroplasmy were significantly correlated in our series. These data suggest that the T8993G mutation is directly responsible for the clinical phenotype, irrespective of the influence of different mitochondrial or nuclear gene backgrounds. On the contrary, no correlation was found between heteroplasmy and biochemical defect in six members of our series (table 2). The L155R amino acid change is thought to alter the structure of the proton channel contained in the membrane bound F0 portion of complex V, thus interfering with the formation of the transmembrane electrochemical gradient. By contrast, ATP hydrolysis, which is what is actually measured in our biochemical assay,15 is carried out by the water soluble portion of the complex, the F1 particle.21 The two portions of mitochondrial complex V are structurally and functionally distinct. It is possible that a defect in the F0 portion of the complex has a limited effect on ATPase function, at least in conditions, such as those of our assay, in which hydrolysis of ATP is measured independently of the presence of the electrochemical gradient.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Linear regression of the logarithm of age of disease onset (in months) versus the percentage of mtDNA heteroplasmy in 10 clinically affected members. The values of heteroplasmy in blood were used in nine cases. In one case (patient II/1, pedigree E) the available value in muscle is reported. R= correlation coefficient.
The availability of controlled systems, such as transmitochondrial cybrid clones, which can yield intact mitochondria with a precisely determined amount of mutant ATPase 6, and the establishment of transgenic systems in normal and ATPase-less Escherichia coli,22 or in other organisms, will help carry out the experimental work necessary to clarify the molecular mechanisms in this disease.
Acknowledgments
We thank the patients and their families for their collaboration, as well as Dr Cremonte, Dr Veneselli, and Dr Scarabino for referring some of the patients and providing diagnostic material. We are indebted to Ms B Geehan for revising the manuscript. Telethon-Italy (grant No 767 to MZ) and ARIN (Associazione Italiana per la promozione delle Ricerche Neurologiche) are gratefully acknowledged for their support. This paper is dedicated to the memory of our friend and colleague Professor Anita E Harding.