Article Text

Abstract
Background: Developmental eye anomalies, which include anophthalmia (absent eye) or microphthalmia (small eye) are an important cause of severe visual impairment in infants and young children. Heterozygous mutations in SOX2, a SOX1B-HMG box transcription factor, have been found in up to 10% of individuals with severe microphthalmia or anophthalmia and such mutations could also be associated with a range of non-ocular abnormalities.
Methods: We performed mutation analysis on a new cohort of 120 patients with congenital eye abnormalities, mainly anophthalmia, microphthalmia and coloboma. Multiplex ligation-dependent probe amplification (MLPA) and fluorescence in situ hybridisation (FISH) were used to detect whole gene deletion.
Results: We identified four novel intragenic SOX2 mutations (one single base deletion, one single base duplication and two point mutations generating premature translational termination codons) and two further cases with the previously reported c.70del20 mutation. Of 52 patients with severe microphthalmia or anophthalmia analysed by MLPA, 5 were found to be deleted for the whole SOX2 gene and 1 had a partial deletion. In two of these, FISH studies identified sub-microscopic deletions involving a minimum of 328 Kb and 550 Kb. The SOX2 phenotypes include a patient with anophthalmia, oesophageal abnormalities and horseshoe kidney, and a patient with a retinal dystrophy implicating SOX2 in retinal development.
Conclusion: Our results provide further evidence that SOX2 haploinsufficiency is a common cause of severe developmental ocular malformations and that background genetic variation determines the varying phenotypes. Given the high incidence of whole gene deletion we recommend that all patients with severe microphthalmia or anophthalmia, including unilateral cases be screened by MLPA and FISH for SOX2 deletions.
Statistics from Altmetric.com
Malformations of the eye, where one or both eyes are absent (anophthalmia) or significantly reduced in size (microphthalmia) are observed in 2–3 per 10 000 births.1 2 Around 60% of individuals with anophthalmia or microphthalmia have systemic malformations, but only 25% of these are part of well defined syndromes.2 Heterozygous mutations in SOX2 are found in up to 10% of patients with microphthalmia or anophthalmia, and SOX2 is therefore the most common causative gene identified to date.3 4 Other genes implicated in such developmental eye anomalies include OTX2,5 PTCH (Gorlin syndrome),6 CHD7 (CHARGE syndrome),7 PAX6,8 RAX/RX,9 CHX1010 and BCOR.11 These ocular malformations are usually associated with haploinsufficiency of the implicated gene, although some missense mutations have been identified.
The phenotype associated with SOX2 haploinsufficiency tends to include severe bilateral eye defects (anophthalmia or microphthalmia with coloboma/cyst) with intrauterine growth retardation, poor growth postnatally associated with pituitary insufficiency, developmental delay including speech and motor problems, seizures, sensorineural hearing loss and male genital abnormalities.4 12–14 MRI findings include periventricular heterotopias, white matter insufficiency, abnormalities of the corpus callosum, hypothalamic-pituitary axis, and hippocampus.4 15–17 Recently mutations in SOX2 have been demonstrated in some cases of anophthalmia-oesophageal-genital syndrome or anophthalmia/microphthalmia and oesophageal atresia (AMEA) (OMIM 600992).18 19
SOX2 is a member of the SOX family of proteins that play a key role in the regulation of embryonic development and are part of a high mobility group (HMG) domain class of transcriptional regulators.20 The HMG domain is highly conserved among SOX proteins and binds to a specific DNA sequence motif inducing bending of the DNA.21 22 The HMG domain is also involved in interaction with other DNA binding proteins to facilitate cell type specific targeting.23 24 Expression of SOX2 is critical for human eye development and also plays a key role in the development of other tissues.25 26
We initially identified SOX2 mutations in a cohort of 100 patients with severe developmental eye anomalies.3 4 We screened a new cohort of 120 patients and identified 6 new cases with intragenic mutations. As deletions of the whole SOX2 gene have been reported in cases with karyotype abnormalities3 27–29 we postulated that cases with a normal karyotype where no mutation had been detected could harbour submicroscopic deletions of SOX2. We therefore carried out multiplex ligation-dependent probe amplification (MLPA) and fluorescence in situ hybridisation (FISH) on patients with no mutations in SOX2 but with anophthalmia or severe microphthalmia, and identified a further six patients with a deletion of all or part of the SOX2 gene. In this paper we present the clinical and molecular details of these 12 new patients.
MATERIALS AND METHODS
Patients with developmental ocular malformations including anophthalmia, microphthalmia and coloboma were ascertained from a specialist clinic at Moorfields Eye Hospital, London or Birmingham Children’s Hospital. After informed consent, peripheral blood samples or cheek swabs were taken. Genomic DNA was prepared using standard techniques.
PCR, sequencing and FISH analysis
PCR amplification of SOX2 genomic DNA was performed in three overlapping fragments, 20 μl reaction volume, 50 ng DNA, 5 mM each primer (table 1), 7.5 μl premix J (Epicentre, Cambridge, UK), and 0.2 μl “failsafe” Taq polymerase (2.5 units/ul, Epicentre). Cycling parameters were 94°C for 7 min followed by 32 cycles at 94°C for 30 sec, 55°C for 30 sec, 72°C for 30 sec. PCR products were sequenced bi-directionally using standard techniques (Big Dye™ terminator, Applied Biosystems, Warrington, UK). Sequencing primers were those used for initial amplification (table 1). FISH analysis was performed using standard methodology. BAC clones RP11-379M20, 43F17, 252O18, 462A6 and 416O18 were selected from the 32 k human tiling path clone set to use as probes for the SOX2 gene and flanking regions (http://www.ensembl.org/Homo_sapiens/cytoview).
MLPA
Dosage analysis was carried out using MLPA30 probes designed specifically for the 5′ and 3′ ends of the SOX2 gene, the probe binding sites being at bases 107–154 and 698–745 respectively (table 1). The probes were diluted to 1.33 fmol/μl, and 1.5 μl of this was used for MLPA in conjunction with a commercially-available MLPA probe mix (CLL1 - P037; MRC-Holland, Amsterdam) to provide controls. The standard protocol of MRC-Holland30 was used reducing the initial DNA denaturation volume to 3.5 μl to allow for the extra 1.5 μl of SOX2 probe mix.
RESULTS
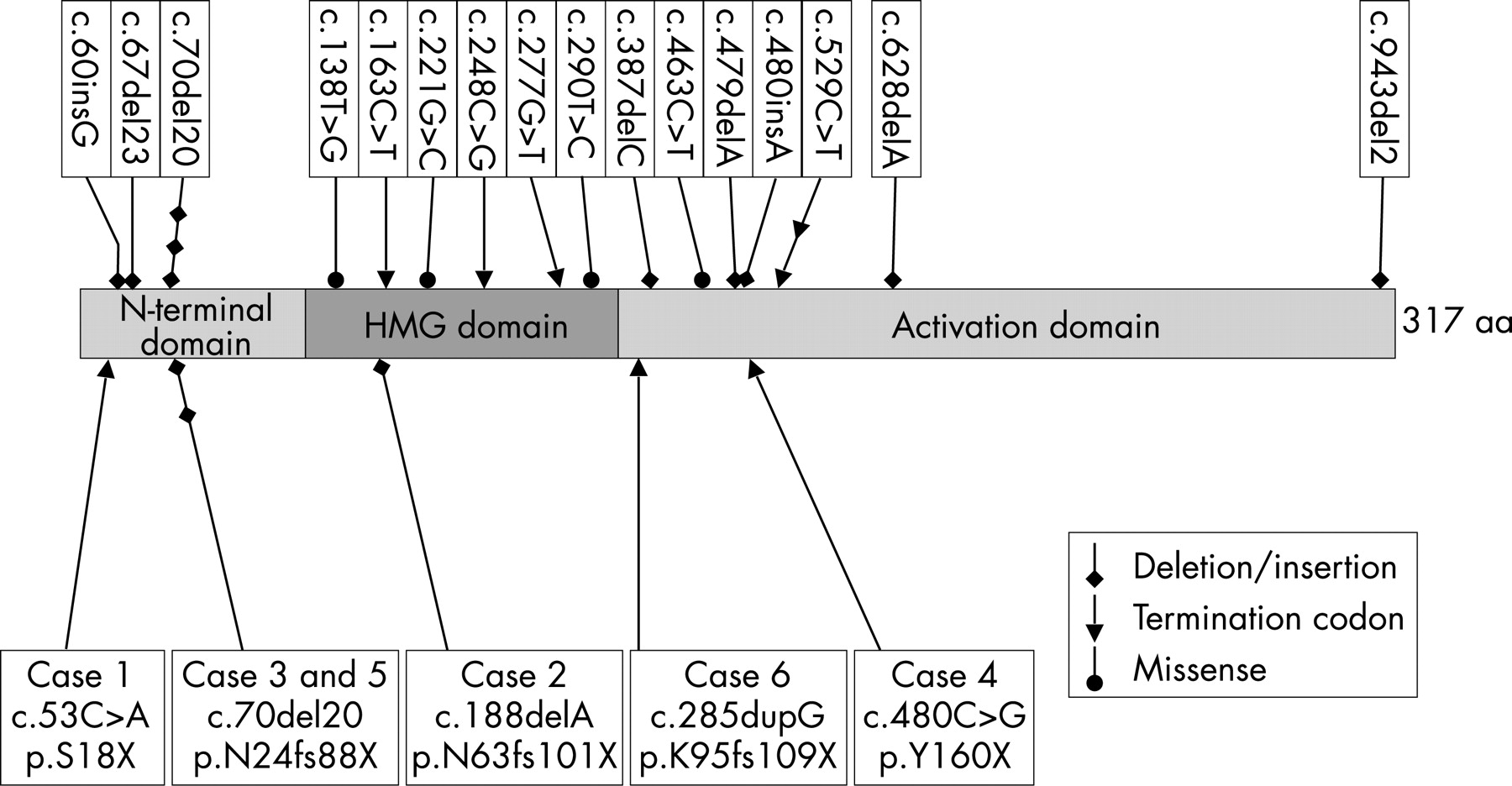
Of the 120 patients analysed, 6 had mutations in the coding region of the SOX2 gene and 7 had a previously reported polymorphic c.976G>A change in the 3′UTR of SOX2. Of the 52 cases that underwent MLPA, five had a deletion of the entire SOX2 gene, two of which were confirmed by FISH and one had a partial deletion that was not resolvable by FISH. The mutations and phenotypes in each case are described below, and in figs 1–3 and table 2. All cases were new.


{kind=link}
{kind=link}
{kind=link}
Case 1 with bilateral anophthalmia is heterozygous for the c.53C>A (p.S18X) mutation resulting in a premature termination codon (PTC), in the extreme N-terminus of the protein, the most 5′ truncation reported for the SOX2 gene. The predicted truncated protein lacks 95% of the protein including the HMG and the C-terminal transactivation domains and leading to likely SOX2 haploinsufficiency. She had fundoplication for gastrooesophageal reflux, delayed motor development, and delayed menarche. She has an unusual gait and shows signs of mild spasticity and dystonia in lower limbs. She has no significant learning disability.
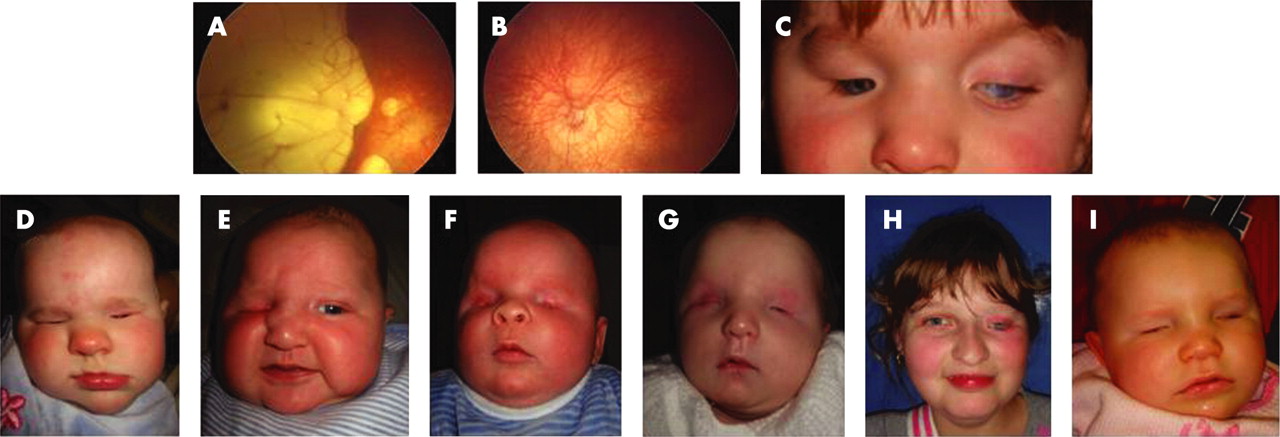
Case 2 with bilateral microanterior segment and atypical coloboma is heterozygous for a single base deletion, c.188delA (p.N63fs101X). This mutation is in the HMG domain of SOX2 and is likely to ablate DNA binding. The predicted truncated protein also lacks the C-terminal transactivation domain. Early problems with vision were suspected postnatally when she had roving eye movements and a right esotropia. The right eye had a reduced (8–9 mm) corneal diameter, eccentric nasally displaced pupil and large atypical colobomatous defects of the retina involving the optic disc and macula with small lacunae around the borders of the main lesion (fig 1). She subsequently developed a dense cataract. The left eye had a normal anterior segment, but the optic nerve was hypoplastic. Both eyes had marked retinal pigmented epithelial hypopigmentation (fig 1A,B), an axial length of 21 mm and 10 D myopia. The electroretinogram showed generalised retinal dysfunction primarily from the inner retina. Her general development was normal for a child with visual impairment. Cranial MR imaging demonstrated corpus callosal thinning.
Case 3 with right severe microphthalmia and left anterior segment dysgenesis and coloboma is heterozygous for the c.70del20 (p.N24fs88X) mutation. The predicted truncated protein lacks the HMG and C-terminal transactivation domains. She had a tracheooesophageal fistula diagnosed in utero and was noted to have additional oesophageal atresia and a horseshoe kidney postnatally. She had delayed motor, speech and cognitive development.
Case 4 with severe right microphthalmia and left anophthalmia is heterozygous for a c.480C>G (p.Y160X) in the transactivation domain. The predicted truncated SOX2 protein lacks three-quarters of the activation domain. Postnatally he had feeding problems and was fed by an NG tube. His right microphthalmia and left anophthalmia were diagnosed at 7 weeks with absent visual evoked potentials. On ultrasound both globes were disorganised and <5 mm axial length. He had a micropenis and cryptorchidism, but his early development was normal.
Case 5 with bilateral anophthalmia is heterozygous for the c.70del20 mutation. No ocular globes were seen on ultrasound. Her cranial MRI scan did not reveal any intracranial malformation and her early development was not delayed.
Case 6 with bilateral anophthalmia has a c.285dupG (p.K95fs109X) mutation in the HMG domain resulting in truncation of SOX2 in the N-terminus of the activation domain. His early development was normal.
Case 7 with bilateral anophthalmia is heterozygous for a deletion of the 3′ end of the SOX2 gene. MLPA analysis using the 5′ probe showed this part of the gene to be present. MLPA with the 3′ probe indicated a hemizygous deletion with the proximal breakpoint between bases 154 and 698 (fig 3). PCR amplification of the whole gene gave no truncated amplicon indicating that the distal breakpoint was outside the gene and the entire gene distal to the internal breakpoint was deleted (data not shown). The deletion was not visible by standard karyotyping and was not detectable with the BAC probes used. She has no dysmorphic features and her development was normal.
Cases 8–12 were all deleted for the 5′ and the 3′ MLPA probes, indicating heterozygosity for deletion of the whole SOX2 gene (fig 3).
Case 8 has right anophthalmia and left cataract. FISH confirmed a deletion of ∼328 Kb with the distal breakpoint within the BAC RP11-462A6 that showed a diminished signal. He was the only case to have seizures and also had brachycephaly, a narrow right nasal passage, cryptorchidism, micropenis considered secondary to isolated gonadotrophin deficiency, delayed speech and overall development.
Case 9 has severe bilateral microphthalmia. FISH analysis was not performed as no material was available. His development was within normal limits for a child with severe visual impairment at 12 months, but he was still not walking at his last assessment at 16 months. He has feeding difficulties. Cranial MR imaging and was normal.
Case 10 has severe bilateral microphthalmia with retinal detachments, but maintains perception of light. FISH studies showed that the deletion is a minimum of ∼550 Kb and extends beyond the most distal probe used in this study (RP11-416O18). She has delayed growth, but otherwise her development is normal.
Case 11 has left anophthalmia with normal right vision with a slightly dysplastic optic disc. She also had ureteric reflux, slightly pointed ears and unusual nostrils. She had delayed motor, speech and cognitive development, and late onset of puberty. FISH studies were unavailable.
Case 12 has right anophthalmia and left microphthalmia with sclerocornea. Her early development was normal. FISH studies were unavailable.
DISCUSSION
We have identified 12 new cases with severe congenital eye defects caused by de novo mutations or deletions of the SOX2 gene. Six of these were total or partial deletions of the gene; two of the five total deletions had genomic deletions extending distal to SOX2. None of the deletions was visible by standard karyotyping. All six intragenic mutation cases generated a PTC. Two had the previously reported c.70del20 mutation16 17 and four were novel mutations: c.53C>A (p.S18X), c.188delA (p.N63fs101X), c.480C>G (p.Y160X) and c.285dupG (p.K95fs109X). As SOX2 is a single exon gene, these mutations are not expected to invoke nonsense-mediated mRNA decay and are therefore predicted to generate truncated SOX2 proteins. Although this could lead to a dominant negative effect, three of these mutations delete all or most of the HMG DNA binding domain (fig 2) and two are deleted for the transactivation domain. Given the similarities in phenotype between these cases and those with a whole gene deletion, it is likely that the truncated proteins are non-functional and result in haploinsufficiency for SOX2.
In addition to the six SOX2 mutation cases described here, 19 further patients have been reported (fig 2).3–5 12–14 16–19 Out of the total of 25 cases, 20 have nonsense or frameshift mutations generating a PTC. An additional frameshift mutation, c.943del2 causes continued translation beyond the normal termination codon and an elongated SOX2 protein.3 The remaining three are missense mutations in the HMG domain. These are likely to disrupt SOX2 function as the HMG domain is highly conserved and is critical for DNA binding and interaction with other proteins. In particular the R74P and L97P mutations are likely to create a bend and change the 3D structure of SOX2.31
There are two mutations that have repeatedly arisen in apparently unrelated individuals. The c.529C>T (p.Q177X) occurred in two patients.3 4 12 The c.70del20 mutation has been described previously in three individuals, although two were twins,16 17 in addition to the two cases described here. A repeat sequence flanking the 20 bp deleted region could be responsible for its recurrence during DNA replication or by unequal recombination as suggested by Zenteno and colleagues.16 17
Interestingly the mutations do not include any C>T transitions at CpG sites, which is unusual for such a CG rich gene as this is the most common point mutation found in mammals. This might reflect the small number of SOX2 mutations so far identified or it might reflect the methylation status of the gene, methylated CpGs being much more predisposed to C>T transition than unmethylated.
All 12 cases described here, and all but 1 of those previously described, are de novo mutations. The exception is one case with c.138T>G (p. N46K) associated with gonosomal mosaicism in an unaffected mother,14 conferring an increased recurrence risk in subsequent pregnancies. However, parental gonosomal mosaicism cannot be excluded for cases where low levels of mutation are not detected in parental blood or in large deletion cases detected by MLPA, although they might be resolvable by FISH. The low incidence of familial mutations could be a reflection of reduced genetic fitness. One cause might be the occurrence of male, and even female genital tract abnormalities (NKR, unpublished data), possibly related to reduced hypothalamic-pituitary-gonadal axis hormones, or direct effect of SOX2 haploinsufficiency on the germ cells.12 In the mouse, SOX2 is expressed in the male and female germ cells, and SOX2 heterozygotes show reduced male fertility, associated with testicular abnormalities.12 25
In keeping with previously documented cases, all 12 patients had a relatively severe ocular phenotype. Nonetheless, the phenotype of case 2 was quite distinct from the majority of reported cases in that both globes were formed, and there was a superior chorioretinal defect involving the optic disc and macula associated with a retinal dystrophy. The only other example of a patient with bilateral formed globes is one of the twins with the c.70del20 mutation.16 17 The retinal dystrophy is particularly interesting in view of mouse studies of retinal development that have shown that precise regulation of SOX2 dosage is critical for the temporal and spatial regulation of retinal progenitor cell differentiation.32 That SOX2 plays an important role in the development of the lens, retina and optic fissure closure is further illustrated by cases 3 and 9 with unilateral cataract; cases 9 and 10 with retinal detachments, and several cases with either iris, or chorioretinal colobomas.
The extraocular phenotype was variable and not determined by mutation type (table 2). Of the two cases presented here with c.70del20, only case 3 had postnatal growth retardation, oesophageal and kidney abnormalities. Comparison with the other three previously reported cases with c.70del20 shows additional variation, even between twins.16 17 Cases 1 (p.S18X), 8 and 11 (whole SOX2 gene deletion) all had pituitary abnormalities. Given the variability, even among patients with the same mutation, it is likely that background genetic or environmental variation has a significant influence. The discordancy in the monozygotic twins is of interest, only one manifesting the ocular phenotype. As the genomic sequence background is identical it cannot be the cause of discordancy, so it could be assumed that environmental or epigenetic factors have had an influence. Epigenetic differences have been identified in monozygotic twins with discordant phenotypes for other conditions.33 34 Other features typical of SOX2 anophthalmia syndrome seen in our cohort included delayed motor and general development, seizures and postnatal growth retardation (<10th centile). We also describe a new phenotype of horseshoe kidney. It is possible that patients with phenotypes such as horseshoe kidney, abnormalities of oesophageal development or retinal dystrophy without classic anophthalmia or microphthalmia could have SOX2 mutations. This possibility is highlighted by the c.70del20 case16 17 with oesophageal atresia and tracheooesophageal fistula, but a normal eye phenotype.
Previous reports have suggested that SOX2 mutations account for 10% of cases with anophthalmia/microphthalmia, this percentage could be higher as we have shown that whole or partial deletion of the SOX2 gene is present in some patients. We used MLPA and FISH to detect these deletions, which would not have been seen in studies specifically targeting intragenic mutations. Such partial gene deletions have been found in other genes,35 but are not identified by standard point mutation analysis methods and might comprise a larger proportion of gene mutations than has hitherto been suspected. The MLPA technique is being increasingly used in routine mutation analysis and will undoubtedly lead to the identification of further small intragenic or partial gene deletions.
In cases with no intragenic mutation or whole gene deletion, as yet undetected deletions close to the SOX2 gene might be present that repress transcription of the gene by position effect, as reported for some other transcription factor genes.36 In conclusion, we have identified 12 new cases of SOX2 anophthalmia syndrome, six of which have whole or partial deletion of the SOX2 gene. We suggest that such deletions could be a relatively common cause of SOX2 anophthalmia syndrome and both tests should be included in the initial diagnostic analysis of anophthalmic/microphthalmic patients. Furthermore, the variability of phenotype exhibited by our patients and other reported cases suggests that patients with horseshoe kidney, oesophageal atresia or tracheooesophageal fistula, deafness, congenital hypopituitary-hypothalamic disorders12 without the classic ocular features of anophthalmia or microphthalmia might also harbour SOX2 mutations. The analysis of a larger number of patients with a wider range of phenotypes is required to try to identify the incidence of such cases and further clarify the range of phenotypes associated with mutation of the SOX2 gene.
Acknowledgments
We thank the referring clinicians and the families for their support and participation in this research. NR is a Senior Surgical Scientist funded by the Academy of Medical Sciences/The Health Foundation (http://www.acmedsci.ac.uk and http://www.health.org.uk, respectively). We are grateful to additional generous support from VICTA (Visually Impaired Children Taking Action) and the Polak Trust. We would like to thank Marie Restori for ultrasound measurements, Dr Graham Holder for electrodiagnostics and the Medical Illustration Dept., Moorfields Eye Hospital for photography.
REFERENCES
Footnotes
Competing interests: None.
Note: PB and DOR should be considered joint first authors.
- Abbreviations:
- FISH
fluorescence in situ hybridisation
- HMG
high mobility group
- MLPA
multiplex ligation-dependent probe amplification