Article Text

Abstract
BACKGROUND Congenital cataract, when inherited as an isolated abnormality, is phenotypically and genetically heterogeneous. Although there is no agreed nomenclature for the patterns of cataract observed, a recent study identified eight readily identifiable phenotypes.
METHODS The Moorfields Eye Hospital genetic eye clinic database was used to identify a four generation family with isolated autosomal dominant congenital cataracts. All individuals (affected and unaffected) underwent a full ophthalmic assessment.
RESULTS The results of the molecular linkage study identifying a missense mutation in the gene encoding the major intrinsic protein of the lens (MIP) have been published elsewhere. Affected individuals had bilateral discrete progressive punctate lens opacities limited to mid and peripheral lamellae with additional asymmetric polar opacification. One young female had predominantly cortical cataract and another had serpiginous nuclear opacities.
CONCLUSIONS This phenotype has not been recorded in human families before and has been termed polymorphic. The pattern of opacification appears to reflect the distribution of MIP in the lens. Furthermore, this is the first clear evidence of allelic heterogeneity in this condition following the identification of a family with lamellar cataracts who have a different mutation within the MIP gene.
- cataract
- linkage
- phenotype
- mutation
- major intrinsic protein
Statistics from Altmetric.com
Inherited cataract is the most common treatable cause of visual loss in children.1 When it occurs in isolation, it is most often transmitted as an autosomal dominant trait.2 Many different phenotypes are recognised but as yet no agreed classification exists. A recent study of families with isolated non-syndromic human inherited cataract3 recorded eight clearly distinguishable phenotypes—anterior polar, posterior polar, nuclear, lamellar, blue dot, coralliform, cortical, and pulverulent.
Inherited cataract is also genetically heterogeneous. Independent chromosomal loci have been mapped to 1p36, 1q21-25, 2q33-35, 13q, 16q22.1, 17p, 17q11-12, 17q24, 21q, and 22q. A recessive form has been linked with the I-blood group locus and characterisation of balanced chromosomal translocations has identified further loci on 2p22.3, 14q24, and Xp (Table 1a). Underlying mutations are now known in the crystallin genes at the loci on 2q,4 21q,5and 22q,6 the connexin genes on 1q7 8 and 13q9 and the developmental regulator genePITX3 on 10q (Table 1b).
(a) Mapped loci for human autosomal dominant congenital cataract, (b) identified human congenital cataract mutations
We have identified a family with autosomal dominant progressive lamellar opacification of the lens associated with anterior and posterior polar cataract. This “polymorphic” phenotype has not previously been recorded in human pedigrees. We have genetically linked this family to the long arm of chromosome 12, to the locus 12q14, and have shown that in this and another genetically unrelated family, a mutation in the MIP gene underlies cataractogenesis.10 In this paper, we describe the clinical spectrum of the disease.
Spontaneously occurring dominant mutations in major intrinsic protein (MIP) have already been shown to produce heritable cataract traits in mice. The CatFr mouse has a splice site mutation that leads to cataract and wrinkling of the capsule. The lop mouse (lens opacity) has a missense mutation which leads to mistargeting of the protein and accumulation in the endoplasmic reticulum of the lens fibre cell.11 The underlying mechanisms by which each results in cataractogenesis remain to be elucidated.
Patients and methods
The genetic database at Moorfields Eye Hospital provided details of the proband, from whom the pedigree was constructed and the family was invited to participate in the study. Ten affected and six unaffected members (Fig 1) underwent ophthalmological examination. Peripheral blood samples were taken for DNA extraction and the linkage study undertaken as previously described.12 Ethics committee approval was obtained for this study and patients gave their written consent to participate.

Abridged pedigree of family with polymorphic congenital cataract.
Results
The results of the molecular linkage study mapping the polymorphic cataract in this family to chromosome 12q14 and the subsequent detection of a mutation in the MIP gene have already been published.10
In the family, autosomal dominant inheritance was supported by the presence of affected male and female individuals in each generation and male to male transmission. The disease showed complete penetrance and highly variable expressivity.
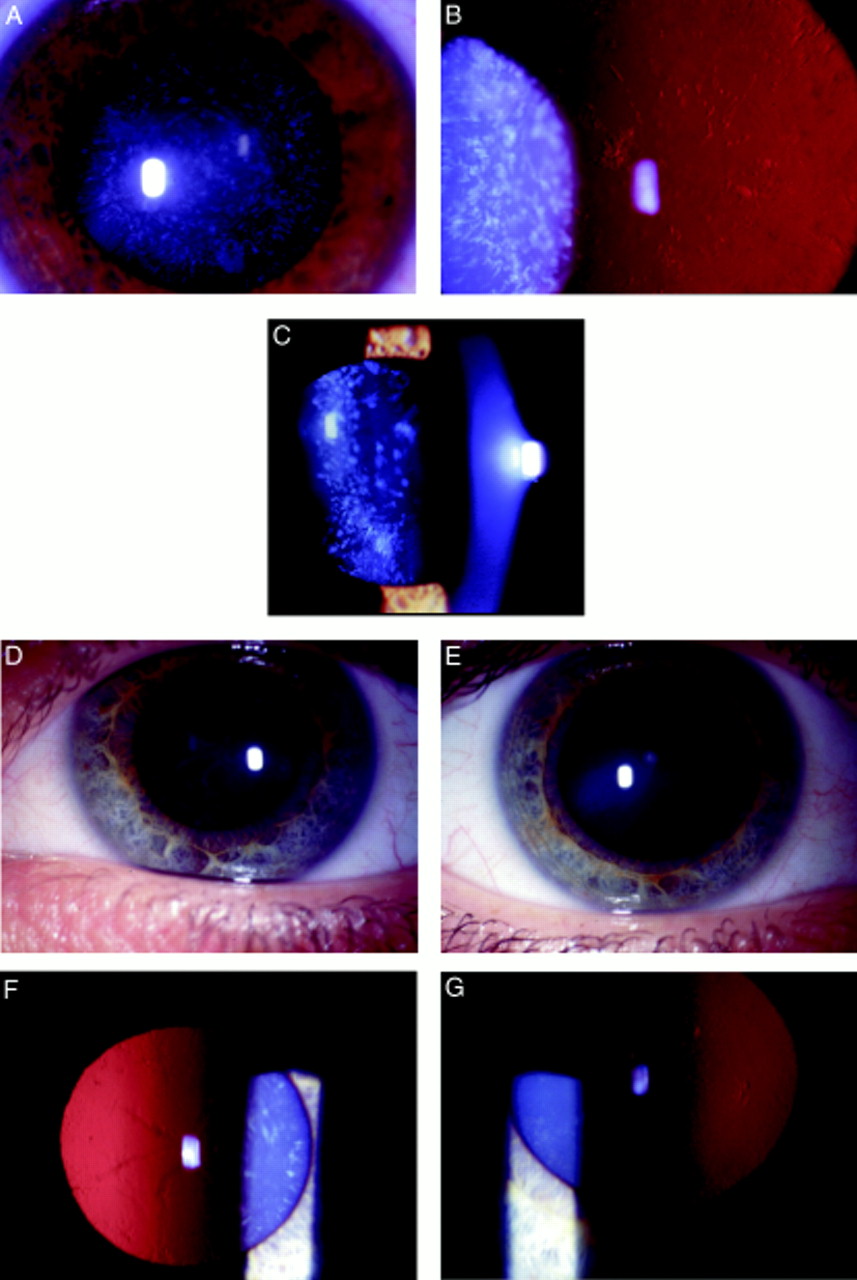
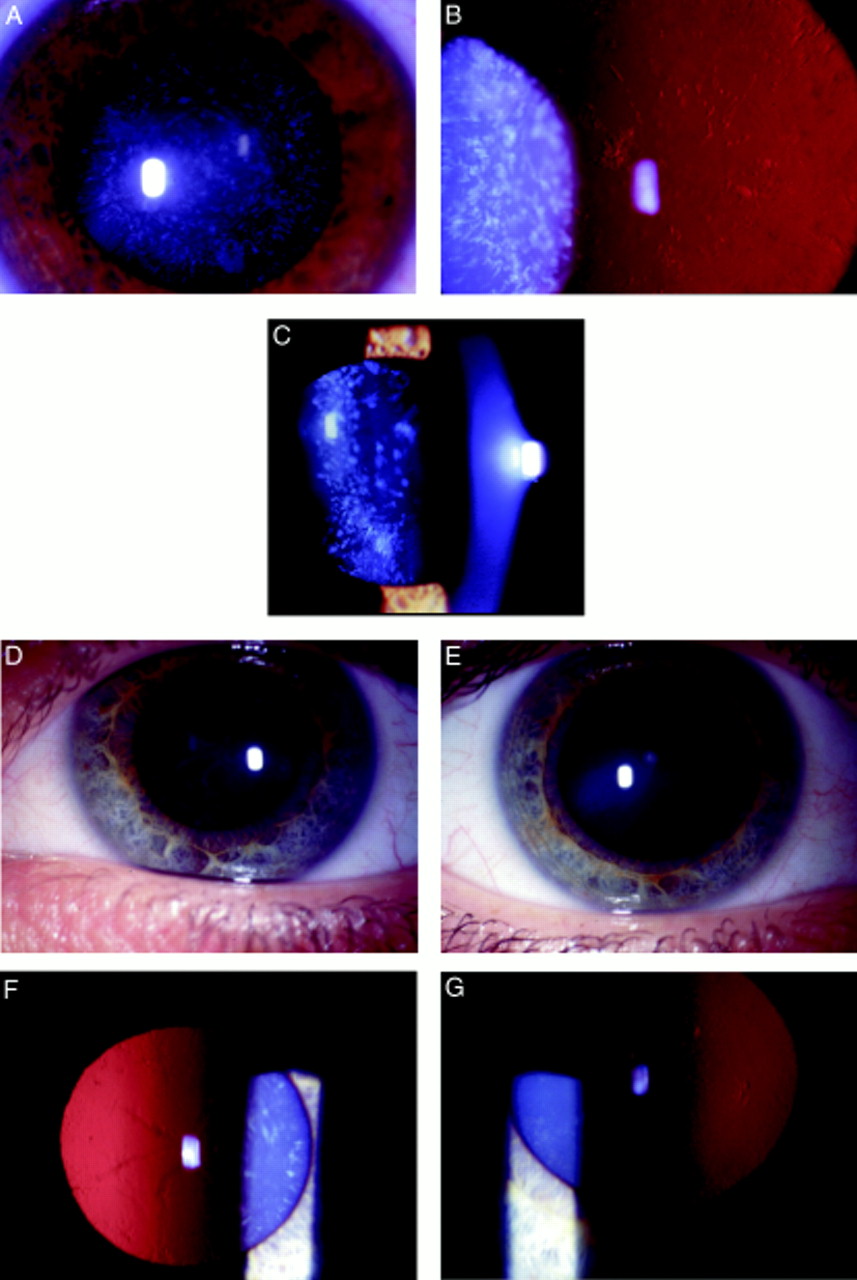
Opacification of the lens was bilateral in all affected cases and consisted of discrete progressive punctate lens opacities limited to mid and peripheral lamellae. In addition, some of those affected had asymmetric anterior and posterior polar opacification. One young female had predominantly cortical cataract and another had serpiginous nuclear opacities. The clinical spectrum of the phenotype is shown in Figure 2. Hospital records indicated that the opacity was present at birth or developed within the first year of life. Visual acuity in the unoperated eyes of those affected ranged from 6/6 to 6/24 (Table 2). No affected individuals have developed strabismus or retinal detachment. There was no evidence of other ocular or systemic abnormalities.
{kind=link}
{kind=link}
Punctate mid and peripheral opacities in: (A) direct illumination (34 year old female); (B) retroillumination (same patient as (A)); (C) slit beam illumination (17 year old female);(D) serpiginous nuclear opacities (24 year old female); (E) anterior polar cataract (same patient as in (D)); (F) retroillumination view of serpiginous nuclear opacities (same patient as in (D)); (G) retroillumination view of anterior polar cataract in patient with predominantly cortical opacification.
Clinical data
Three individuals had undergone bilateral lensectomy in childhood, aged 1 month (left and right best corrected visual acuities 6/9, 6/12 respectively), 4 months (6/6, 6/9; subsequently developed well controlled aphakic glaucoma), and 15 years (HM, 6/9). Right extracapsular cataract extraction had been performed on two patients in adulthood (aged 55 and 72 years, each achieving best corrected visual acuity of 6/6).
Discussion
In this paper, we document a new inherited cataract phenotype resulting from a mutation in the gene that encodes the major intrinsic protein of the lens (MIP), the most abundant membrane protein in the mature lens fibre cell.
A number of roles have been suggested for MIP but it is now known to be one of a number of sequence related proteins known as aquaporins, which form channels to selectively transport water molecules across cell membranes.13 The missense mutation we have described substitutes an amino acid thought critical for this specialised function.
Ten mammalian aquaporins are now known.14 Often more than one is expressed in a single tissue. The proteins are suggested to have important functions in the eye, being involved in aqueous humour dynamics and lacrimation. Aquaporins also have significant roles in renal water reabsorption, cerebrospinal fluid secretion, and the generation of pulmonary secretions have been suggested.15
To date, only aquaporin-2 has been implicated in human disease. It is found abundantly in kidney collecting duct epithelium, where its function is regulated by ADH (antidiuretic hormone). Several missense mutations (recessive and dominant) have been identified that result in the development of nephrogenic diabetes insipidus.16Interestingly, polymorphisms of aquaporin-1 (present on the red blood cell membrane, the Colton blood group antigens) have been documented that produce a non-functional protein (the null phenotype) but whose effects are subclinical.17 Such evidence has suggested that other mechanisms are able to compensate for aquaporin dysfunction. Our evidence that mutations in aquaporin-0 underlie cataract formation confirm the critical role of these proteins in certain tissues.
The phenotype of the cataract in this family has not previously been observed as an inherited trait18 and to reflect the variable appearance between eyes and among individuals has been termed “polymorphic”.
The discrete punctate opacities are similar to the pulverulent phenotype, which is characterised by fine dust-like opacification. However, the opacities in this pedigree are larger and, critically, their position within the lens is consistent contrasting markedly with the variable distribution of pulverulent opacities. Polar opacification is also not a feature of the pulverulent phenotype.
The phenotype observed in our family is also clearly distinct from the incompletely penetrant non-progressive polymorphic cataract mapped to chromosome 2q33-35 by Rogaev et al 19 in the Turkmen populations of the former Soviet Union. In this cataract opacities resembled lumps of grapes or cotton anywhere from the fetal nucleus to the equator. Polar opacification was also not observed.
Although the mature lens fibre cell is metabolically inert, aquaporin water transport would be expected to continue as it is not energy dependent. Thus, the progressive nature of the cataract might well be explained as the cumulative effect of cellular dysfunction over time. The presence of polar cataract is less easily explained but parallels the distribution observed in the CatFr mouse.
Screening our panel of other families with isolated inherited congenital cataract identified a second pedigree with a different dominant missense mutation in the MIPgene.10 Lens opacification in this family was non-progressive and confirmed a perinuclear lamella. Such an observation is intriguing because it is not clear why two missense mutations in similar parts of the gene would produce such strikingly different phenotypes. Furthermore, this provides the first confirmation of allelic heterogeneity in this condition.
In conclusion, we have reported the identification of a new congenital cataract phenotype that results from a missense mutation in the major intrinsic protein of the lens. Studies of the implications of the mutation for the function of the protein will provide further understanding of how the underlying genotype relates to the phenotype observed.
Acknowledgments
The authors thank the family for their participation in this project and Mr Philip Ball (senior medical illustrator, Addenbrooke's Hospital, Cambridge) for his help in preparing the illustrations. This work was supported by a grant from the Wellcome Trust, 053416 to ATM and SSB.