Article Text

Abstract
Aims: To describe the clinical phenotype of X linked juvenile retinoschisis in eight Italian families with six different mutations in the XLRS1 gene.
Methods: Complete ophthalmic examinations, electroretinography and A and B-scan standardised echography were performed in 18 affected males. The coding sequences of the XLRS1 gene were amplified by polymerase chain reaction and directly sequenced on an automated sequencer.
Results: Six different XLRS1 mutations were identified; two of these mutations Ile81Asn and the Trp122Cys, have not been previously described. The affected males showed an electronegative response to the standard white scotopic stimulus and a prolonged implicit time of the 30 Hz flicker. In the families with Trp112Cys and Trp122Cys mutations we observed a more severe retinoschisis (RS) clinical picture compared with the other genotypes.
Conclusion: The severe RS phenotypes associated with Trp112Cys and to Trp122Cys mutations suggest that these mutations determine a notable alteration in the function of the retinoschisin protein.
- Italian families
- X linked juvenile retinoschisis
- XLRS1 gene
- mutations
- phenotype
Statistics from Altmetric.com
Congenital retinoschisis is a rare bilateral vitreoretinal disorder characterised by vitreous degeneration and splitting of the retina between the nerve fibre and ganglion cell layers. The patients have typically a cystic-like stellate maculopathy or a foveal schisis,1 and in 50% of cases bilateral inferotemporal retinoschisis.1,2 The electroretinogram is beneficial in the diagnosis of juvenile retinoschisis. The scotopic amplitudes are more severely affected than the photopic amplitudes. The a-wave can be of normal or reduced amplitude in this disorder,3 whereas the amplitude of the b-wave is appreciably reduced,4,5 giving a negative wave tracing.
The disease is transmitted as an X linked recessive trait, occurring almost exclusively in males, although a few affected female carriers have also been identified, some having a family history of consanguinity,6,7 supporting homozygosity of X locus.
The cloned retinoschisis gene (XLRS1) maps to the distal short arm of the X chromosome (Xp22).8 The expression is restricted to the photoreceptors and bipolar cells9–11 and the protein contains a conserved region found in other proteins that participate in cell-cell interactions.8
Some reports have been published describing the clinical features in families with defined mutations in the XLRS1 gene.7,12–15
In this study we describe six different mutations, two of which have not been previously described, in the XLRS1 gene and we determine the clinical phenotype associated with these genotypes in eight Italian families.
PATIENTS AND METHODS
Five families with X linked retinoschisis (XLRS) diagnosed at the department of ophthalmology, Seconda Università di Napoli, one at the department of ophthalmology, Università “Federico II” di Napoli, and two XLRS families diagnosed at the department of ophthalmology HSR, Università di Milano, Italy, were included in this study (Fig 1). The research procedures were carried out in accordance with institutional guidelines and the Declaration of Helsinki. Informed consent was obtained from all patients and controls after the nature of the procedures was fully explained.
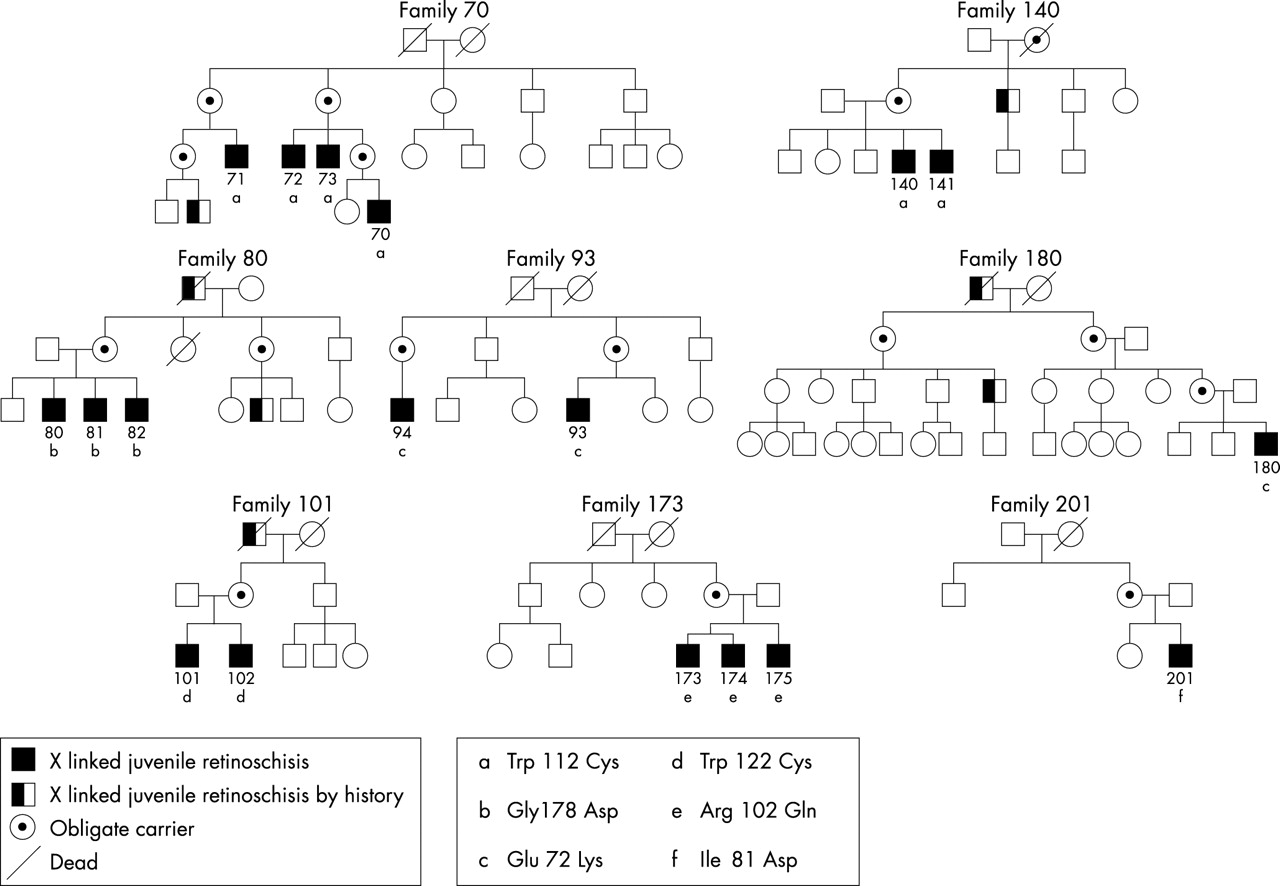
Pedigrees of families with X linked juvenile retinoschisis and identified mutations in the XLRS1 gene.
Complete ophthalmic examinations were performed in 18 affected male (mean age 23 (SD 9) years) and 14 female (mean age 40 (SD 6) years) obligate carriers. The clinical examinations included best corrected visual acuity with Snellen visual chart, slit lamp biomicroscopy, fundus examination, fundus photography, fluorescein angiography, A and B scan, standardised echography, and electroretinography.
The electroretinogram (ERG) was recorded by means of corneal contact lens electrodes with a Ganzfield stimulator according to international clinical standards.16 The ERG results from the XLRS patients were compared with those of 20 control subjects with normal vision, whose ages ranged from 15–33 years; we defined “reduced” as values below mean −2 SD of controls values.
The diagnostic criteria in XLRS1 male patients included macular abnormalities defined as typical foveal schisis, blunted foveal reflex, pigmentary demarcation lines or retinal pigment atrophy with or without peripheral retinoschisis, reduced ERG b-wave, and a history of bilateral visual impairment since childhood.1,17
Blood samples from 18 retinoschisis patients belonging to eight families were collected. Genomic DNA was isolated from lymphocytes by standard methods. The coding sequences of the XLRS1 gene were amplified by polymerase chain reaction (PCR) and directly sequenced on an automated sequencer (ABI 3100; Applied Biosystem, Foster City, CA, USA) using the ABI-PRISM big dye terminator cycle sequencing ready reaction kit (Applied Biosystem). The pathogenic effect of the mutations identified was confirmed by excluding their presence in 100 normal X chromosomes.
RESULTS
Six different XLRS1 mutations were identified (Table 1). Similar to previous reports,18 all the mutations were missense mutations and were clustered in exons 4, 5, and 6 encoding the discoidin motif. To the best of our knowledge, two of these mutations have not been previously described—namely, the Ile81Asp and the Trp122Cys. Both these mutations affect amino acids that are highly conserved in the discoidin motif across evolution, changing them to residues never found at this position in related proteins.18
Identified mutations in the XLRS1 pedigrees
The clinical data of patients are reported in Table 2. The mean age at the onset of disease in the 18 XLRS1 male patients was 5.6 (SD 2.3) years and the symptoms at the onset were photophobia and/or hemeralopia, nystagmus, and reduced visual acuity, but in a few cases the patients were asymptomatic and diagnosed after ophthalmological screening. Best corrected visual acuity varied from 20/20 to light perception (mean values 20/60 (SD 20/100)); the refractive spherical errors ranged from −4.50 to +5 dioptres (mean values −0.1 (SD 2.3)). Hypermetropia in our patients, unlike in the reports of other studies,19–21 was not the most frequent refractive error; the mean value of the axial length calculated with immersion standardised A-scan echography was 22.9 (SD 0.91).22
Clinical data of XLRS1 families
The vitreous abnormalities observed ophthalmoscopically were vitreous veils in 18 eyes (51.4%), vitreoretinal tractions in 18 eyes (51.4%), and falciform folds in three eyes (8.6%). All patients who underwent A and B-scan standardised echographic examination showed vitreoschisis in both eyes, with the hyaloid adherent to the underlying retina, while two patients, who had previously undergone eye surgery for retinal detachment, showed posterior vitreous detachment, a probable consequence of surgery.23
Macular abnormalities were present in all affected patients; 27 eyes (77.1%) showed a typical foveal schisis, a cystic-like stellate alteration, five eyes (14.3%) showed macular atrophy and in three eyes macular coloboma, macular scarring, and healthy macula were present, respectively.
Macular abnormalities were not related to any genotype. The presence of bone spicule pigmentation was evident only in pedigree 140 but not in pedigree 70 with the same mutation.
Peripheral retinoschisis was evident in the temporal sector in 17 of the 35 eyes studied (48.6%) and 10 of these (58.8%) also showed retinal detachment; the families with Trp112Cys, Trp122Cys, and Arg102Gln mutations showed peripheral retinoschisis.
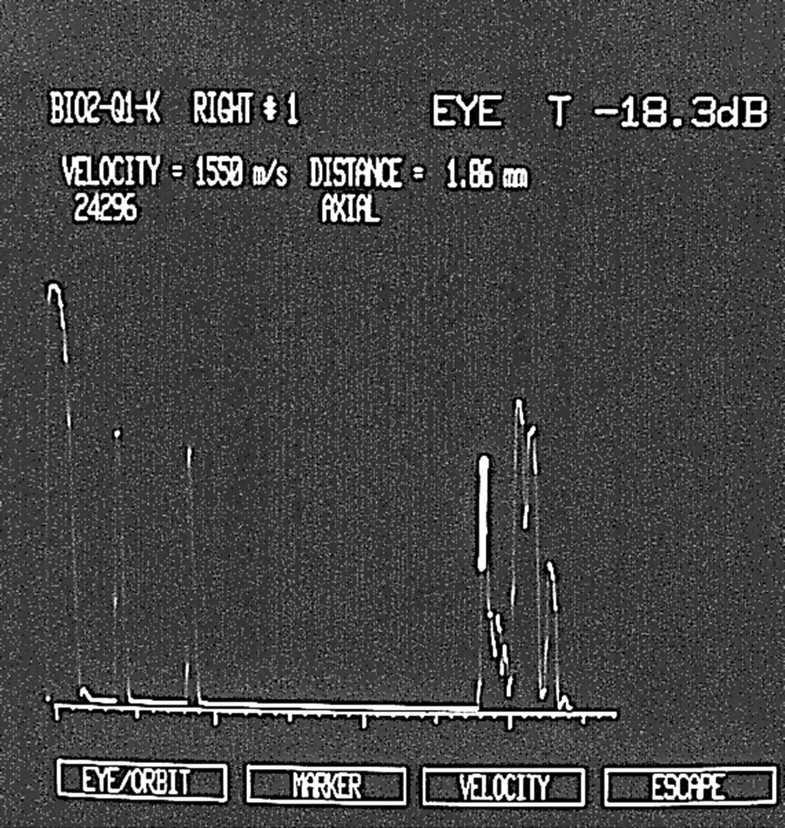
Moreover, in all patients we noted an increased thickness of the retinal choroid layer in the macular region (2 (SD 0.2) mm; in normal controls, the value is <1.5 mm).24 This increased macular thickness (Fig 2) is due not only to macular schisis, but also to a thickening of the choroid. The increased thickness of the choroid is present only in the posterior pole.

{kind=link}
{kind=link}
Echographic examination recorded in an affected XLRS male, showing an increased thickness of retinal choroid layer in the macular region.
Electroretinography was performed for 11 patients (Table 3), as five patients refused the examination and two patients showed an ERG performed in another eye clinic before ocular surgery for retinal detachment. In all pedigrees the typical response to white single flash was seen with a reduction of the b-wave amplitude and a relative preservation of the a-wave amplitude, causing a reduced b/a ratio. The b/a ratio was reduced (<1.1) in nine patients (81.8%) while two patients (18.2%) had a reduction in both the a and b-wave amplitudes, with a normal b/a ratio. Moreover, the amplitude of the a-wave was reduced in four patients and this reduction did not appear to be related to the duration of the disease. These patients showed a Trp112Cys mutation of the XLRS1 gene. The scotopic blue flash response was reduced in seven patients (<275 μV) and extinct in four patients. The photopic b-wave amplitude was reduced in eight patients (<124 μV) while three patients had extinct responses. The genotype of the patients with extinct scotopic or photopic ERG was Trp112Cys. The 30 Hz flicker amplitude was reduced in three patients (<34 μV) and extinct in the patient with Glu72Lys genotype. The implicit time of the 30 Hz flicker was prolonged in six patients.
Electrophysiological data of XLRS1 families
DISCUSSION
A study of the genotype/phenotype correlation in XLRS is complicated because of the low number of XLRS1 patients and because most mutations take place within the discoidin domain, which probably results in a similar effect on the function of this protein.
Although a great number of mutations have been identified in the XLRS1 gene,18,25–28 there are few clinical data relating to the different genotypes.7,12–15
This is the first study reporting a genotype-phenotype correlation on XLRS1 in Italian patients in which we found two mutations not previously described.18
In Glu72Lys XLRS1 families, we showed typical foveal schisis without peripheral schisis and the presence of vitreoschisis and vitreous veils in all patients, something not described in the families studied by Scinoida.14
The phenotype associated with the Gly178Asp genotype showed a similar clinical picture in brothers, with foveal schisis, vitreous veils, vitreoretinal tractions, vitreoschisis, and peripheral schisis. Moreover, in this pedigree, in accordance with what has been reported by Sieving13 in a XLRS1 subject of 80 years of age with the same mutation, the ERG findings showed the classic alteration of the inner retina with a sparing of the a-wave function, resulting in the reduction of the b/a ratio to the maximal ERG response.
In the families with Trp112Cys and Trp122Cys mutations we observed a more severe RS clinical picture compared with the other genotypes. The Trp112Cys families showed a typical foveal retinoschisis with peripheral retinoschisis, vitreoretinal tractions, retinal detachment, and a severe reduction of the retinal function, as evident from the reduced amplitude of both the a and b-waves to the maximal scotopic ERG and from the extinct scotopic and photopic tracing in most of the subjects, despite the same duration of the disease as the other families. The phenotype associated with the novel mutation Trp122Cys, which is typical foveal retinoschisis with peripheral schisis, also showed a severe phenotype, as evident from the need for surgery for retinal detachment and from the marked alteration in the electrophysiological results performed 2 years before surgery which suggested extensive retinal impairment, not consistent with congenital retinoschisis.
No clinical phenotype is distinguished but we note that some ERG changes are associated with certain mutations. In fact, Trp 112 Cys is associated with very poor b-wave amplitudes (4/4 patients) and reduced a wave amplitudes in 2/4 patients tested. Trp 122 Cys is associated with reduced b and a wave amplitudes in 2/2 patient tested. Other mutations showed the more classical reduced b-wave with preservation of the a-wave (Table 3).
The severe XLRS phenotypes associated with Trp112Cys and Trp122Cys mutations suggest that these mutations determine a dramatic alteration in the function of the retinoschisin protein. The tryptophan residues involved in these mutations are highly conserved in other discoidin domains,18 indicating that these two amino acid residues have an important role in the function of this domain and, consequently, of the retinoschisin protein. It is also important to note that the replacement of a large hydrophobic residue such as tryptophan with a smaller and more hydrophilic residue such as cysteine will result in a corresponding alteration in the protein structure of the discoidin domain. Since this domain is a protein-protein interaction module, it is conceivable that a disruption will impair the ability of retinoschisin to interact properly with its putative partners yet to be identified.
The molecular elucidation of X linked juvenile retinoschisis may serve as a key to understanding the pathogenesis and, perhaps, provide a better tool for use in clinical diagnosis, prognosis, and genetic counselling.
Acknowledgments
This study was supported by grant law 41 (1999) from the Regione Campania and by the Telethon Foundation.