Article Text

Abstract
AIMS To determine the different morphologies of autosomal dominant cataract (ADC), assess the intra- and interfamilial variation in cataract morphology, and undertake a genetic linkage study to identify loci for genes causing ADC and detect the underlying mutation.
METHODS Patients were recruited from the ocular genetic database at Moorfields Eye Hospital. All individuals underwent an eye examination with particular attention to the lens including anterior segment photography where possible. Blood samples were taken for DNA extraction and genetic linkage analysis was carried out using polymorphic microsatellite markers.
RESULTS 292 individuals from 16 large pedigrees with ADC were examined, of whom 161 were found to be affected. The cataract phenotypes could all be described as one of the eight following morphologies—anterior polar, posterior polar, nuclear, lamellar, coralliform, blue dot (cerulean), cortical, and pulverulent. The phenotypes varied in severity but the morphology was consistent within each pedigree, except in those affected by the pulverulent cataract, which showed considerable intrafamilial variation. Positive linkage was obtained in five families; in two families linkage was demonstrated to new loci and in three families linkage was demonstrated to previously described loci. In one of the families the underlying mutation was isolated. Exclusion data were obtained on five families.
CONCLUSIONS Although there is considerable clinical heterogeneity in ADC, the phenotype is usually consistent within families. There is extensive genetic heterogeneity and specific cataract phenotypes appear to be associated with mutations at more than one chromosome locus. In cases where the genetic mutation has been identified the molecular biology and clinical phenotype are closely associated.
- autosomal dominant cataract
- linkage
- phenotype
- lens
Statistics from Altmetric.com
Infantile cataract is one of the commonest treatable causes of childhood blindness in the USA and Europe1 with a prevalence of 1–6 cases per 10 000 births.2 Inherited cataract accounts for up to half of all congenital cataract3 and the commonest mode of inheritance is autosomal dominant. There is considerable phenotypic variation in autosomal dominant cataract (ADC) but there have been few systematic studies of the different morphologies. Loci for the genes causing autosomal dominant cataract have now been identified across the human genome (Table 1).
Autosomal dominant cataract loci
In 1910 Harman3 described the appearance of the lens opacities seen in families with inherited cataract and listed five groups—lamellar, coralliform, stellate, anterior, and posterior polar, and finally an “undefined” group. Clapp later divided congenital cataract of all aetiologies into two broad groups, complete and partial, and then subdivided the partial cataracts into zonular, coralliform, punctate, discoid, pyramidal, Y opacities, floriform, fusiform, disc-shaped, and Morgagnian.4Duke-Elder,5 François,6 and Vogt7 have described the wide variety of phenotypes seen in individuals with inherited cataract but have not studied the pedigrees affected by ADC. We have undertaken a study of the clinical appearance of the lens in a large number of families with dominantly inherited cataract and have been able to identify a number of distinct phenotypes and to correlate some of these with the chromosomal location of the disease mutations.
Patients and methods
We examined 161 affected individuals with ADC from 16 pedigrees. The families were drawn from the genetic clinic database at Moorfields Eye Hospital and had no other ocular or systemic abnormalities. All patients underwent an eye examination with particular attention to the lens and cataract morphology. Families were included if there were three generations of affected individuals with at least four affected members. Anterior segment photographs were taken where possible and peripheral blood was collected for DNA extraction to be used in subsequent linkage studies. Ethical approval for the study was given by the hospital ethics committee and informed, signed consent was obtained from the patients. In subjects where surgery had been performed early, a description of the phenotype was taken from the hospital notes.
Genomic DNA was extracted from EDTA sequestered blood samples and linkage analysis using Généthon microsatellite markers8 was performed using a previously described protocol.9
Results
Eight phenotypes could be recognised that included all the pedigrees seen in the study. These phenotypes were anterior polar, posterior polar, nuclear, lamellar, coralliform, blue dot (cerulean), cortical, and pulverulent (Table 2). The morphology of the cataract in affected individuals within any single pedigree was the same for all except the pulverulent pedigrees, varying only in the severity and density of the opacity. In pedigrees with the pulverulent phenotype there was considerable variation in the cataract morphology among affected individuals from the same pedigree. Male to male transmission was seen in 12 families. In the remaining families, the severity of affected members was equal in both males and females suggesting autosomal dominant inheritance.
Pedigree details
ANTERIOR POLAR CATARACT
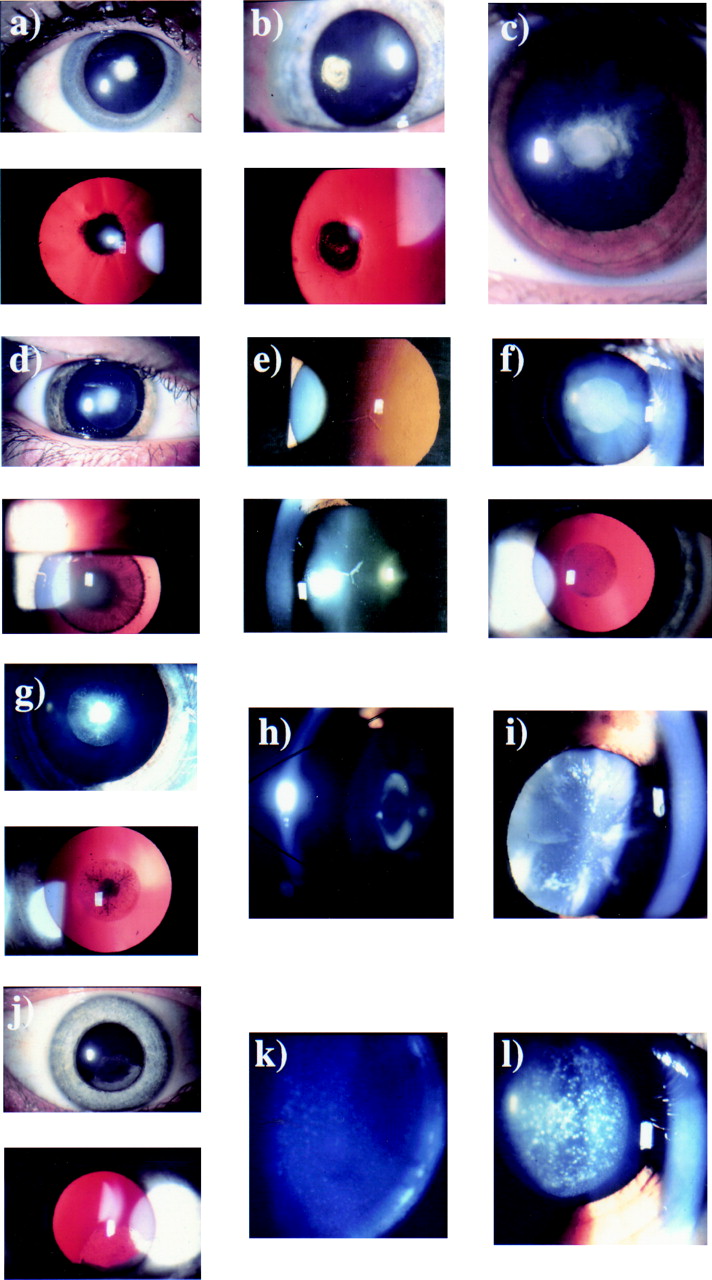
Anterior polar cataracts are situated at the anterior pole of the lens and have been reported to have a minimal effect on visual acuity.10 11 We studied a single family with anterior polar cataract. Thirty family members (excluding partners) were examined and 19 found to be affected. Seven patients had undergone surgery and the phenotype was examined in the remaining 12 cases. The opacity was always in the same position of the lens, the anterior pole, but varied in severity.12 The larger opacities were pyramidal with a base of 4 mm and extended into the anterior chamber (Fig 1a), while the smaller opacities consisted of a flat plaque measuring under 1 mm in diameter. In one affected individual who showed the disease haplotype the only sign of an anterior polar abnormality was an irregularity of the anterior capsule. We have demonstrated linkage of anterior polar cataract to chromosome 17p in this family13 showing significantly positive lod scores for markers D17S849 (Zmax= 4.01, θmax= 0.05) and D17S796 (Zmax= 4.17, θmax= 0.05). Multipoint analysis gave a maximum lod score of 5.2 between these two markers.
{kind=link}
Autosomal dominant congenital cataract phenotypes. (a) Slit lamp and retroillumination view of anterior polar cataract (47 year old woman). (b) Slit lamp and retroillumination view of a stationary posterior polar cataract (49 year old woman). (c) Slit lamp view of a progressive posterior polar cataract (9 year old female). (d) Slit lamp and retroillumination view of a dense nuclear cataract (14 year old male). (e) Slit lamp and retroillumination view of nuclear opacities (50 year old woman). (f) Slit lamp and retroillumination view of the Coppock-like cataract with fine nuclear opacities (24 year old woman). (g) Slit lamp and retroillumination view of the Coppock-like cataract with dense central opacities (6 year old female). (h) Slit lamp view of lamellar cataract. (i) Slit lamp view of a blue dot (cerulean) cataract (32 year old man). (j) Slit lamp and retroillumination view of a cortical cataract (45 year old woman). (k) Slit lamp view of a fine pulverulent cataract (32 year old man). (l) Slit lamp view of large pulverulent opacities (12 year old female).
POSTERIOR POLAR CATARACT
In the pedigrees studied the posterior polar opacity was always at the posterior pole of the lens. Two types of posterior polar cataract were identified in this study—a stationary posterior polar cataract and a progressive posterior polar cataract. The stationary posterior polar cataract consisted of a single well defined posterior polar plaque (Fig 1b), which varied in size among individuals as well as between the eyes of an individual. No other part of the lens was affected and although we have not studied the family over a prolonged period, the clinical records suggest that the cataract did not increase significantly in size. In a pedigree affected by this stationary posterior polar cataract 15 members of the family were examined and 10 found to be affected. Seven patients had undergone surgery and details of the cataract were obtained from the notes in four of these cases, all of whom were described as having posterior polar cataract.
We have demonstrated linkage of this phenotype to chromosome 1p14 with significantly positive lod scores for markers D1S508 (Zmax= 3.14, θmax= 0) and D1S468 (Zmax= 2.71, θmax= 0). Multipoint analysis gave a maximum lod score of 3.48 (θmax= 0.07) between markers D1S508 and D1S468.The second family with posterior polar cataract was affected by a progressive posterior polar cataract that initially consisted of a posterior plaque but later developed posterior cortical extensions (Fig 1c) with punctate cortical opacities causing a progressive deterioration of visual acuity requiring cataract extraction. Twenty one family members were examined, 11 were affected and all but one unilateral case had undergone surgery. Clinical records available in nine of the 10 operated cases documented the progressive nature of the cataract; the average age of diagnosis was 5 years but surgery was performed at a mean age of 20 years (range 3–40 years, median 19 years, mode 16 years). We have excluded linkage of this phenotype to the loci known to be associated with ADC.
NUCLEAR CATARACT
Nuclear cataract consists of opacities confined to the embryonic and fetal nuclei of the lens. The cataract varies such that some patients have dense central opacities surrounded by fine dots (Fig 1d), while others have fine dot opacities distributed throughout the embryonic and fetal nuclei (Fig 1e), with opacification of the anterior and posterior Y sutures. The opacities are confined to the fetal nucleus. Two pedigrees affected by nuclear cataract were examined. In the first family 21 individuals were examined and 12 found to be affected. Of these, six had undergone surgery and the clinical notes were available in two cases. Preliminary linkage results in this family have excluded the loci known to be associated with ADC.
In the second family affected by nuclear cataract 19 family members were examined and 11 found to be affected. Eight patients had undergone surgery and in seven of these the clinical notes gave an accurate description of the cataract morphology. The cataracts in three unoperated patients were examined. The mildly affected individuals had dust-like opacities throughout the embryonic and fetal nuclei (Fig 1f) while the more severely affected patients had a dense central opacity surrounded by a haze of fine powdery opacities (Fig 1g). We have demonstrated linkage of this pedigree to the γ crystallin locus on chromosome 2q (D2S157 Zmax= 5.28, θmax= 0.00) but have discovered that this pedigree is related to the Coppock-like pedigree initially studied by Smith15 and Harman16 and previously linked to the γ crystallin gene.17 The Coppock-like cataract phenotype is well described in the literature but no photographs of the opacity have ever been published (Fig 1f and g).
LAMELLAR CATARACT
Lamellar cataract is also referred to by some authors as zonular cataract and is one of the commonest phenotypes of ADC. The inner fetal nucleus is made up of clear lens surrounded by an opacified shell which is in turn surrounded by clear cortex (Fig 1h) which may contain opacities referred to as “riders” or “cortical spokes”. Lamellar cataract represents a disturbance in the lens development at a particular time and the cataractous “shell” varies in size according to the stage of fetal development at which the disturbance occurs. The cataract may be mild and the visual acuity may be well preserved or it may be dense enough to need surgery in childhood. Three families affected by lamellar cataract were studied. One of the families consisted of 14 members of whom eight were affected. All underwent surgery although one patient had unilateral cataract extraction. The surgery was carried out at a mean age of 7 months (range 2–36, mode 2, median 2). The individual who still had a cataract in one eye has a lamellar cataract with a visual acuity of 6/60 but has refused surgery. In the rest of the family details of the phenotype were restricted to clinical records that were available in four cases. In the other two families all the affected individuals had undergone surgery. In one family of 14 people there were nine affected individuals and the notes were available in two cases. In the third family there were also 14 members but with eight affected individuals and notes available in three cases. All were described as having lamellar cataract. These pedigrees have yet to be the subject of linkage studies.
CORALLIFORM CATARACT
The single pedigree of this phenotype from the Moorfields genetic data base is descended from a family originally described by Nettleship.18 The name of this phenotype is derived from the resemblance of the cataract to sea coral,19 having finger-like tubular protuberances extending from an opacified nucleus. Nineteen family members (excluding partners) were examined and eight found to be affected. All affected members of this pedigree had had surgery and the description of the cataract was taken from the clinical notes that were available in seven cases. The mean age at operation was 5 years (range 1–12 years, median 5, mode 7 years). This pedigree has not yet been the subject of a linkage study.
BLUE DOT (CERULEAN) CATARACT
The blue dot cataract first described by Vogt20 is not believed to be congenital but develops in childhood, progresses slowly, and does not require surgery until adulthood.21 22 The opacities are distributed throughout the lens, with the outer nucleus containing blue-white dots that become more numerous in the cortex where they may form large cuneiform (wedge-like) shapes in the mid-periphery (Fig 1i). In this pedigree seven of 13 patients were diagnosed as having the cataract and five of the affected seven were examined. The cataract was diagnosed at a mean age of 17 years and patients underwent surgery at a mean age of 27 years (range 10–47, mode 25, median 25). The phenotype was consistent within the family. This pedigree has been excluded from the two loci already known to be associated with blue dot (cerulean) cataract. The blue dot locus on chromosome 17 was excluded with the markers D17S1790 (−2.46 at θmax= 0.05), D17S802 (−3.40 at θmax= 0.05), D17S1847 (−2.66 at θmax= 0.05), D17S836 (−3.04 at θmax= 0.05), and D17S784 (−1.27 at θmax= 0.05). The blue dot locus on chromosome 22 was excluded with the single marker D22S315 (−2.63 at θmax= 0.05).
CORTICAL CATARACT
Cortical cataract is rarely described as an autosomal dominant trait. In the family studied as part of this project the cataract presented in childhood or early adult life and varied in severity but not in morphology. The nucleus was unaffected and the opacified fibres were restricted to the outer cortex in contact with the lens capsule affecting a sector of lens (Fig 1j) and being different in this respect from lamellar cataract. The pedigree we studied consisted of 20 family members of whom only six were affected with the cataract and four of these were available for examination. In three of the affected members the cataract affected only the superior cortex, the nucleus remained unaffected, and there was no opacity elsewhere in the lens. Nine eyes of the six affected patients had undergone surgery which was performed at a mean age of 23 (range 7–43). This pedigree has yet to be the subject of a linkage study.
PULVERULENT CATARACT
Pulverulent cataracts are characterised by powdery (pulverised) opacities that may be found in any part of the lens. Pedigrees affected by pulverulent cataract showed a wide intrafamilial variation in the cataract phenotype and, although some affected individuals from these families had cataracts similar to lamellar or nuclear cataract, pedigrees were classified as pulverulent if the majority of affected individuals had fine, powdery opacities (Fig 1k). Five families had pulverulent cataract. In one of the families 23 members were examined and 16 found to be affected. The cataract varied between affected individuals. In some the fine opacities formed a lamellar distribution with a clear cortex and minimal inner fetal nuclear involvement. Others had more widely spread pulverised opacities extending into the cortex with no demarcation of the fetal nucleus. In some the cataract was mild and the fine dust-like opacities were clustered only around the anterior and posterior Y sutures. In this first pedigree nine individuals had undergone surgery at a mean age of 17 years (range 3–82, median 8, mode 11). This pedigree has been linked to chromosome 13 with a maximum two point lod score of 4.06 (θmax= 0.00) with marker D13S175.9 In a second family with pulverulent cataract 15 members were examined and 10 were affected. Four had undergone bilateral cataract surgery, two unilateral, and four had not had surgery. The mean age at surgery was 14 years (range 5–22 years, median 17). The cataract had powdery opacities throughout the lens although they were more abundant in the fetal nucleus. In some the opacities were large and coarse (Fig 1l) and in others finer and dust-like.
A third family affected by pulverulent cataract consisted of 15 family members of whom eight were affected by the cataract. Only two patients had undergone surgery and examination of the remaining six revealed fine dust-like opacities predominately in the fetal nucleus but also in the cortex, with moderate opacification of the Y sutures.
The fourth family affected by pulverulent cataract consisted of 21 family members with eight affected individuals. The phenotype in this family showed great intrafamilial variation, some had the typical fine dots throughout the lens, others had posterior opacities with the rest of the lens unaffected, and one individual had dense bilateral nuclear opacification.
A fifth family, previously shown in 1963 to cosegregate with the Duffy blood group locus on chromosome 1q was recontacted and 18 members of the pedigree (10 affected, six unaffected, and two spouses) examined. All affected individuals had previously undergone cataract surgery, their hospital records indicating that dense opacification was present at birth. Linkage to 1q has been confirmed (maximum multipoint lod score 5.22 at θmax= 0) and a missense mutation identified in the gap junction α8 gene that encodes connexin 50.23
Discussion
In this study of ADC we have demonstrated that the phenotypes of the pedigrees encountered could be classified as one of the eight following types—anterior polar, posterior polar, nuclear, lamellar, coralliform, blue dot (cerulean), cortical, and pulverulent. This study was limited to large families that were suitable for linkage studies but this was thought not to affect the distribution of the affected phenotypes. Other phenotypes not encountered in this study have been reported but they have either been described in individual cases and not in families or are very uncommon. Although an attempt was made to examine the phenotypes of all family members at risk of inheriting the affected gene, the description of intrafamilial variation in the phenotypes was limited by the number of patients who had already undergone surgery. This is a limitation inherent in all studies of early onset inherited cataract and we attempted to obtain as detailed a description as possible from the hospital records in such cases.
In all the pedigrees apart from the pulverulent pedigrees the morphology was the same in all affected family members but varied in severity. In the pulverulent pedigrees the phenotype varied among affected individuals from the same family. Intrafamilial clinical heterogeneity in ADC has been described by many authors24 25 including interocular variability in individuals affected by ADC.26 The pedigrees described as having this wide variation in morphology are characteristic of the pulverulent type described here. A total of 14 pedigrees have now been mapped to 13 different loci identified as containing genes that give rise to cataract (Table 1). In this study we have assigned a chromosomal locus in four pedigrees and 11 remain to be linked. An anterior polar pedigree maps to chromosome 17p,13 a posterior polar pedigree to 1p,14 a nuclear pedigree now known to be related to the Coppock-like pedigree maps to the γ crystallin region on chromosome 2q, and a pulverulent pedigree maps to chromosome 13.9 We have excluded linkage of a pedigree affected by the blue dot cataract to the two loci known to give rise to this phenotype and have also excluded linkage of a posterior polar pedigree and a pulverulent pedigree to the known cataract loci. Further linkage studies are under way.
Clinically similar phenotypes of autosomal dominant cataract map to different regions of the genome and different phenotypes map to the same locus: two unrelated pedigrees affected by blue dot (cerulean) cataract have been mapped to different chromosomes27 28and two unrelated pedigrees affected by posterior polar cataract have also been mapped to different chromosomes.14 29 Two different phenotypes, a posterior polar14 and the Volkmann cataract,24 both map to what may prove to be the same locus on chromosome 1p. In contrast, in families where the underlying genetic mutation has been identified the resultant phenotype correlates with the underlying molecular biology of the lens: genes for human crystallin proteins have now been mapped and are important candidate genes for ADC formation. The Coppock-like cataract is a nuclear cataract caused by a mutation in the γ crystallin gene complex on chromosome 2q30 and the γ crystallins are predominantly found in the lens nucleus.31 The blue dot (cerulean) cataract consists of multiple blue-white dots that predominate in the lens cortex and β crystallins are more abundant in the outer (cortical) layers of the lens. A pedigree affected by blue dot (cerulean) cataract has been demonstrated to have a mutation in the β crystallin region on chromosome 22q28 with the affected members having a chain termination mutation in the human β crystallin gene CRYBB2.32 A missense mutation in the gene for connexin 50, a gap junction membrane protein, on chromosome 1q has been shown to give rise to pulverulent cataract in humans.23
A missense mutation in the human α crystallin gene CRYAA has been shown to give rise to a series of eye abnormalities including zonular central nuclear opacities, cortical and posterior subcapsular cataract, microcornea, and congenital microphthalmia.33 The α crystallin protein is found in many tissues in the eye and throughout the body and a mutation in this protein may be expected to have a considerable effect on ocular development.
In mouse models for ADC, mutations in the crystallin and membrane bound proteins have also been shown to give rise to cataract, although the relation between genotype and phenotype is not as clear. ThePhilly mouse has a nuclear and subcapsular cataract34 caused by a mutation in one of the β crystallin genes35 and the Elomouse, which is a model for microphthalmia secondary to abnormal lens formation36 is caused by a mutation in one of the γ crystallin genes.37 A mutation in the gene for the major intrinsic protein (MIP) of the lens fibre cell membranes has been implicated as causing the cataracts in both theCat[Fr] mouse and theLop mouse.38 39 The human gene for MIP has been mapped to chromosome 12q40 41 but has not yet been implicated in human cataract.
Although in humans the major crystallins are the α, β, and γ crystallins, contributing 90% of the water soluble proteins of the lens,42 there are many other species specific crystallins that may be relevant to human cataract formation. These crystallins are enzymes that in some cases have been shown to have a different function outside the lens but have been recruited during evolution to perform as lens crystallins owing to their stable structures and/or in some cases their original enzyme activity.43 44 The Volkmann and the posterior polar cataract both map to a region on chromosome 1p within which lies the τ crystallin/α enolase gene. This crystallin is not known to be expressed in the human lens. Indeed, no mutation has been found in the gene in these families and the single family described with red cell enolase deficiency does not have cataract. However, it is tempting to speculate that an abnormality in the gene may be associated with cataract in humans.45
This study of lens morphology in ADC has demonstrated wide clinical heterogeneity. There is clearly considerable genetic heterogeneity and the identification of the causative mutations will allow for a more detailed comparison between the precise mutations and the clinical phenotype as well as offering further insight into the mechanism of human cataractogenesis.
Acknowledgments
We thank the families for agreeing to participate in this study and the UK HGMP Resource Centre for microsatellite primer synthesis and the use of computing facilities. The work was supported by a grants from the Wellcome Trust (043073 to AS and SSB; 053416 to ATM and SSB). ACWI was supported by a grant from The Friends of Moorfields Eye Hospital and DSM is a Wellcome Prize student.