Article Text

Abstract
Background/aims: Patients with autosomal dominant optic atrophy (ADOA) are genetically heterogeneous, but all have disc pallor. A degree of cupping in ADOA can make the distinction from normal tension glaucoma (NTG) clinically difficult. This study aimed to clarify the features of the optic nerve of patients with ADOA at the OPA1 locus.
Methods: 29 patients (58 eyes), from 12 families, were identified in a prospective observational study of patients with ADOA examined by a single observer between 1995 and 1998, in whom genetic analysis showed either evidence for linkage to chromosome 3q28 or mutations in the ADOA gene, OPA1. All of the patients had disc and fundal photographs available for retrospective analysis. Clinical data collected included disc appearance, intraocular pressure, Snellen visual acuity, Hardy-Rand-Rittler colour vision plates, and Humphrey 30-2 visual fields.
Results: Mean age at time of examination was 37 years and mean visual acuity was 6/24. Disc morphology showed temporal disc pallor in 30 eyes (52%) and total disc pallor in 28 eyes (48%). At least one disc showed a cup to disc ratio of more than 0.5 in 18 patients (28 discs, 48%). The temporal neuroretinal rim always showed pallor and shallow shelving (or saucerisation) was seen in 46 eyes (79%). Only 12 discs (21%) had deep excavation and baring of blood vessels. All of the patients had normal intraocular pressure and no family history of glaucoma. There was a temporal grey, pigmentary crescent in 12 patients (18 eyes, 31%) and peripapillary atrophy in 20 patients (40 eyes, 69%), but disc margin haemorrhages were not seen. There was no maculopathy or retinopathy.
Conclusion: The optic disc morphology, described for the first time in this genetically homogeneous population of patients with OPA1 ADOA, shows a distinctive absence of a healthy neuroretinal rim and shallow saucerisation of the optic disc cup, with frequent peripapillary atrophy.
- autosomal dominant optic atrophy
- OPA1
- phenotype
- optic disc cupping
Statistics from Altmetric.com
A utosomal dominant optic atrophy (ADOA), Kjer type1 is an inherited primary optic neuropathy that leads to reduced visual acuity. It presents with an insidious onset of variable visual loss, optic nerve pallor, centrocaecal visual field scotoma, and colour vision deficit.2 Histopathological studies suggest that the underlying defect is due to retinal ganglion cell degeneration. Genetic linkage studies localised a dominant optic atrophy gene (OPA1; OMIM1655003) to chromosome 3q28-qter,4 and we and others have identified the OPA1 gene.5,6 It is a dynamin related GTPase5,6 which is widely expressed, with high abundance in the retina. More than 66 mutations have been reported.5–11 These mutations include missense and nonsense alterations, deletions, and insertions. As over 50% of the mutations reported probably represent null alleles, dominant inheritance of the disease may result from haploinsufficiency of OPA1 protein, although there is also some evidence for a pathophysiological dominant negative effect.11,12 Analysis of the N-terminal leader peptide of the deduced protein sequence suggests the OPA1 gene product is targeted to mitochondria and may exert its function in biogenesis and stabilisation of mitochondrial membrane integrity. Thus, OPA1 ADOA may be due to impairment of mitochondrial function caused by mutations in the nuclear gene OPA1.
The optic nerve appearance in ADOA may range from complete atrophy, through temporal pallor, to subtle pallor. We previously described a clinical group of ADOA patients in whom 55% showed subtle or temporal pallor and 44% had total atrophy of the optic nerve.13 The appearances of the optic disc are usually symmetrical. Best colour vision and least field loss have been noted in patients with the least degree of clinical optic atrophy.13 The visual field defect in ADOA is centrocaecal, central, or paracentral, and often appears as a large defect in individuals with severe disease. The colour vision defect in ADOA may be an acquired tritanopia,1,14 or a generalised non-specific dyschromatopsia.15 Families with red-green colour defects16–18 have been described. Our assessment of patients from families that show evidence of linkage to the dominant optic atrophy locus on chromosome 3q 28-qter has shown that no patient examined displayed a truly isolated loss of tritan discrimination,19 and what is more, there was even intrafamilial variation in colour axis discrimination. Many patients show an absent or delayed pattern visually evoked potential (PVEP),20 showing that there is a conduction defect in the optic nerve. The pattern electroretinogram (PERG) shows an abnormal ratio of waveforms (namely, the N95:P50 ratio), with reduction in the amplitude of the N95 waveform.20–23 Since the PERG N95 component is thought to be specific for the retinal ganglion cell,24 this finding supports a ganglion cell origin for ADOA. Magnetic resonance imaging of the optic nerves in patients with dominant optic atrophy shows a reduced optic nerve sheath complex throughout the length of the intraorbital optic nerve with no signal abnormality and a clearly visible cerebrospinal fluid (CSF) space.25
There may be uncertainty in making the clinical diagnosis in some patients, based solely on the appearance of the optic disc. It is our experience that patients have occasionally been misdiagnosed as suffering from Leber’s hereditary optic neuropathy or normal tension glaucoma.
We undertook a detailed assessment of the disc appearances in patients with ADOA in whom disc and fundal photographs were available for retrospective analysis. All the selected patients were from pedigrees in which either linkage analysis showed evidence of linkage to chromosome 3q28 or which harboured disease associated mutations in the ADOA gene, OPA1.11
PATIENTS AND METHODS
Patient selection
Criteria for inclusion of patients in this study were (1) availability of disc and fundal photographs for retrospective analysis; (2) prospective clinical examination data by a single observer; and (3) family data either showing linkage to chromosome 3q28-qter markers or a documented mutation in the OPA1 gene.
Clinical and photographic examination
Patients were identified from a prospective observational study of patients with ADOA examined by a single observer between 1995 and 1998. Probands from the families with autosomal dominant optic atrophy were identified from the Moorfields genetic clinic database. The clinical criteria for the diagnosis of ADOA and inclusion of families in the study were optic nerve pallor, reduced visual acuity, abnormal colour vision, and a centrocaecal visual field defect in at least one member of the family over 6 years of age, with evidence of autosomal dominant inheritance. In addition to the prospective descriptions of the disc made by the single observer, patients had disc and fundal photographs available for retrospective analysis.
Clinical data collected included disc appearance from clinical examination and photographic survey, intraocular pressure, Snellen visual acuity, Hardy-Rand-Rittler colour vision plates, and Humphrey 30-2 visual fields.
The optic nerve was classified as either normal or showing temporal pallor or total atrophy. The morphology of the optic nerve was defined by cup:disc ratio, neuroretinal rim, excavation, presence of a grey pigmentary crescent, and peripapillary atrophy. The nerve fibre layer abnormalities visible with the direct ophthalmoscope and the 90 dioptre (Volk aspheric) lens with red-free light were noted. A number of patients had scanning laser ophthalmoscopy imaging of the optic nerve head and nerve fibre layer.
The study was approved by the ethics and scientific committee of Moorfields Eye Hospital and written informed consent was obtained prospectively from participants.
Genetic linkage analysis and OPA1 mutation detection
The documented OPA1 mutations in the patients included in this study are reported in a separate publication.9,11 In summary, blood samples were taken and genomic DNA extracted using the Nucleon II kit (Scotlab Bioscience; Strathclyde, Scotland, UK). PCR products were amplified from patient DNA using primers located in flanking intron and UTR sequences.7 All 28 coding exons of OPA1 and intron/exon splice sites were screened for mutations by SSCP, heteroduplex analysis and direct sequencing in one affected individual from each family. Putative mutations were tested for segregation in the respective families by SSCP/heteroduplex analysis or direct sequencing, and in 100 control chromosomes (for methods see Thiselton et al11).
RESULTS
Patients
Fifty eight patients from 12 pedigrees with linkage or mutation data were identified (Fig 1), and of these clinical disc photographs were available on 29 patients (50%). The availability of disc photographs was apparently random, and was chiefly determined by time of day at examination, availability of photographer, and patient consent (Fig 2). Where available the electrodiagnostic features and magnetic resonance imaging of the optic nerve and chiasm have been previously reported.20,25

Twelve pedigrees with OPA1 ADOA described in this study. Solid symbol represents affected individual, open symbol represents unaffected individual. Square = male; circle = female. Tangential line = deceased. *DNA sample analysed. ∼ clinical examination.
{kind=link}
{kind=link}
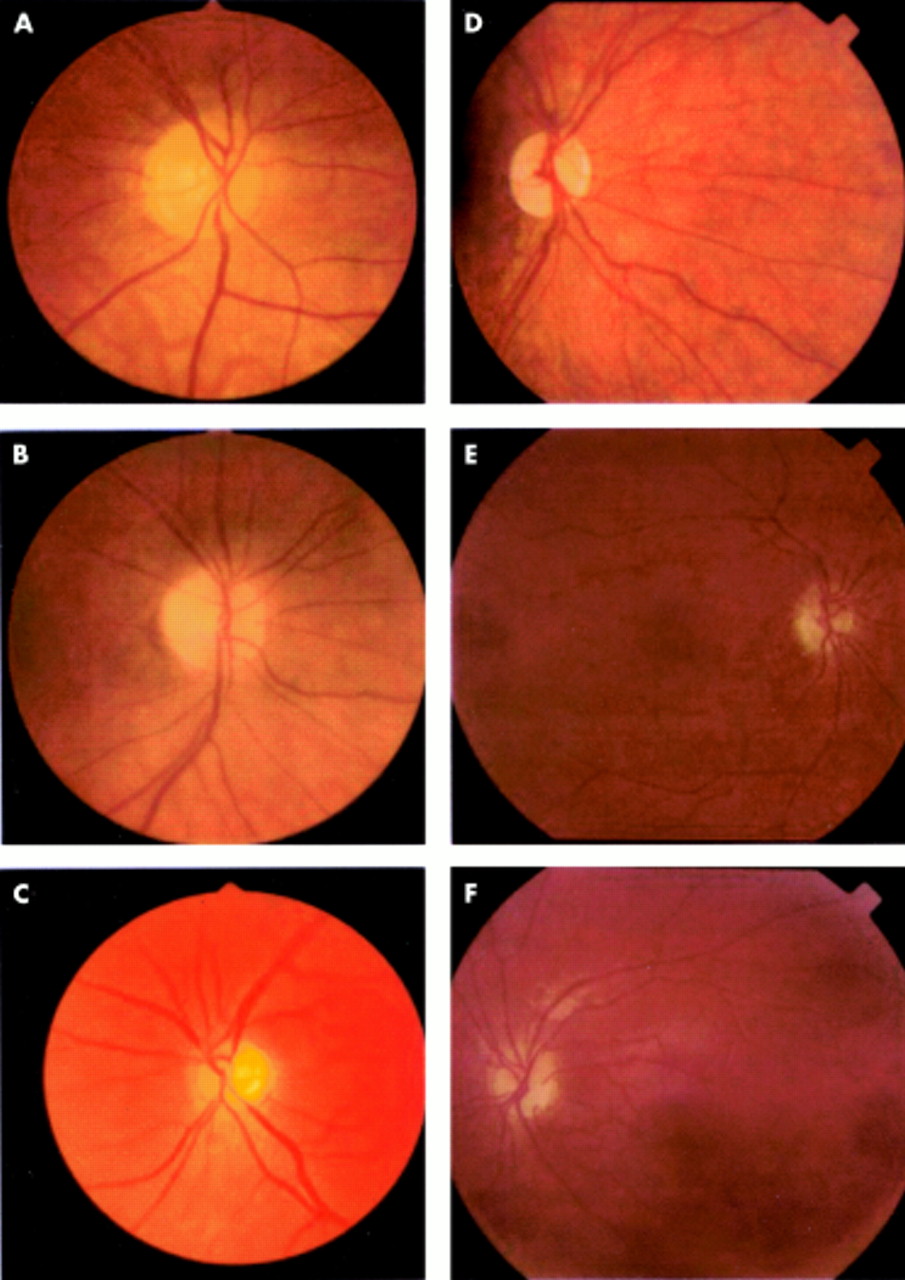
Colour photographs of the optic discs of patients with autosomal dominant optic atrophy in this study illustrating key features: (A) temporal pallor (pedigree B1: IV-15, right eye), (B) total atrophy (pedigree L1: V-2, right eye), (C) optic disc with CDR >0.5 (pedigree C5: II-3, left eye), (D) temporal pigmentary crescent (pedigree T1: III-1, right eye), (E) and (F) peripapillary atrophy (pedigree H1: II-1. right (E) and (F) left eyes).
Clinical profile
The mean age at time of examination was 37 years and mean best corrected visual acuity was 6/24, with a spread from 6/9 to hand movements. There were 13 males and 16 females. The mean age of onset of symptoms was 7 years, with a range of 2–25 years. Intraocular pressures were all within the normal range (<21 mm Hg), with a range of 12–20 mm Hg (mean 14.6 (SD 1.8) mm Hg). Colour vision, assessed by Hardy-Rand-Rittler colour plates for congenital and acquired colour vision deficits showed a tritanopia in 28% (eight patients) and a mixed deficit in 72% (21 patients). Humphrey 30-2 automated perimetry data were available in 24 patients. They showed relative preservation of peripheral visual field in all patients, a centrocaecal scotoma in 34 eyes (71%), and a large central defect in 14 eyes (29%). The clinical features are summarised in Table 1.
Optic nerve features of 29 patients from 12 families with OPA1 ADOA in whom clinical examination data and review of disc photographs were carried out
Of the 12 families, two had a history of affected individuals who had been diagnosed as suffering from Leber’s hereditary optic neuropathy in the past (F3: IV-2 and C7: II-3).
Optic nerve appearances
Disc morphology showed temporal disc pallor in 30 eyes (52%) and total disc pallor in 28 eyes (48%). At least one disc showed a cup to disc ratio of more than 0.5 in 18 patients (total of 28 discs, 48%). Temporal neuroretinal rim showed pallor in all patients. Shallow shelving, or saucerisation, in at least the temporal half of the disc was commonly observed (46 eyes, 79%) and only 12 discs (21%) had deep excavation and baring of blood vessels. Overall, 28 discs (48%) had a cup to disc ratio >0.5. All of these patients had normal intraocular pressure and no family history of glaucoma. Red-free photographs were available in six patients and qualitative red-free observational data was recorded for 19 patients. This showed a thinned or absent retinal nerve fibre layer in the temporal region, corresponding to the papillomacular bundle, in all eyes. Five patients had recorded scanning laser ophthalmoscopy images of the disc and macula. In these patients the NFL was diffusely thinned, particularly in the temporal region of the macula (data not shown), but none of these patients showed a slit-like deficit, in contrast with findings in patients with primary open angle glaucoma.26
There was a temporal pigmentary, or grey, crescent in 12 patients (18 eyes, 31%) and peripapillary atrophy in 20 patients (40 eyes, 69%). Disc margin haemorrhages were not seen in any of the patients. There was no maculopathy or retinopathy.
Chromosome 3q28-qter linkage data and OPA1 mutations
Table 1 shows the mutations in the individuals described in this study,11 or linkage data to chromosome 3q28-qter. A mutation in OPA1 in pedigree H1 was not detected by the means used, but linkage analysis confirmed linkage to the OPA1 locus (maximum Lod score of 3.91). The promoter region of OPA1 has not characterised and was not therefore screened, but further experiments to detect a mutation are ongoing.
Genotype-phenotype correlation
No correlation was detected between the morphology of the optic disc and the type of mutation in OPA1 or to the OPA1 protein domain which would be predicted to be affected. The 12 discs (21%) which showed deep excavation and the 28 discs (48%) which had a cup to disc ratio >0.5 were found in patients with OPA1 mutations throughout the gene.
DISCUSSION
This study of the optic disc and neuroretina in this group of 29 genetically homogeneous patients with OPA1 ADOA refines the diagnostic criteria used in making the clinical diagnosis. The patients described here are from 12 families who all show either evidence of genetic linkage to chromosome 3q28 or who have been shown to have mutations in the OPA1 gene. This is the first reported study of the detailed features of the optic disc in a genetically confirmed population of patients, who all have OPA1 ADOA. Previous studies of disc excavation in ADOA27 have been retrospective and have used a clinical definition of ADOA, but have not investigated or distinguished the genetic aetiology of the optic atrophy. It is therefore possible that patients with a dominant inheritance pattern of optic atrophy, described in other studies, may have been genetically heterogeneous. The visual defects reported in the one published family with ADOA mapping to the locus OPA4 (chromosome 18q12.2–12.328) and the one reported family mapping to OPA2 (Xp11.4-Xp11.229), overlap considerably with that described for ADOA at the OPA1 locus.11,13 The recent identification of mutations in the OPA3 gene found in a recessive syndromic optic atrophy (optic atrophy plus syndrome, or Costeff optic atrophy syndrome30), further demonstrate the genetic heterogeneity that is possible in optic atrophy, especially if a positive history of dominant inheritance is lacking. In addition, it is increasingly recognised that normal tension glaucoma may be dominantly inherited.31,32 Therefore, this study of a genetically homogeneous group of patients is important for observations and conclusions about the morphology of the optic disc in OPA1 ADOA.
The finding of pallor with disc excavation, in the absence of raised intraocular pressure is a feature of a range of optic neuropathies. These non-glaucomatous conditions include arteritic anterior ischaemic optic neuropathy, congenital optic nerve anomalies such as coloboma, methanol toxicity, anterior and posterior non-arteritic ischaemic optic neuropathy, optic neuritis, traumatic and compressive optic neuropathies (for example, tumours and aneurysms affecting the anterior visual pathways).33 What appears to be most characteristic about the optic disc in OPA1 ADOA is the absence of a healthy temporal neuroretinal rim and the shelved appearance of the excavation.
The universal finding of a grey, pigmented temporal crescent within the neuroretinal rim tissue of the optic disc was reported by Fournier et al27 in nine patients with dominant optic atrophy. It is not clear what this morphological feature may represent. We did not find a grey crescent universally in this study. A grey crescent was clearly visible in 18 eyes (31%). A grey, pigmentary crescent around the optic disc was first described by Shields,34 who found that it was more common in African-American patients (12% of normal patients in the reported study) and in myopic patients. All of the patients reported in this paper were white and the highest degree of myopia was −1.25 dioptres.
Peripapillary atrophy in patients with ADOA was seen in this study in 69% of eyes. A concordance of peripapillary chorioretinal atrophy with visual field defects that accompany glaucoma development has been noted in patients with ocular hypertension.35 The extent and location of visual field abnormalities that develop in hypertensive eyes with progression to glaucoma was found to exhibit a concordance with the extent and location of progressive peripapillary atrophy noted in the hypertensive.36
Neither the grey, pigmentary crescent nor peripapillary atrophy are strictly correlated with the severity of the optic disc pallor, although the data suggest that there may be a trend to peripapillary atrophy with increased pallor of the disc.
ADOA caused by mutations in OPA1 may be a mitochondrial disease, albeit with still very little known of the actual mechanisms of disease.5,6 Conservation between OPA1 and its yeast orthologues, Msp1 in fission yeast37 and Mgm1 in budding yeast,38 allows speculation regarding OPA1 pathophysiology and its role in mitochondrial fusion and fission.39 The reported histopathology documents that there is loss of retinal ganglion cells, particularly from the papillomacular bundle and a thinning of the optic nerve substance.40,41 There is also some loss of myelin seen. This loss of myelin could be a primary or a secondary event.42 However, a loss of myelin could lead to secondary changes in the pigmentation seen at the optic nerve head. The chronic appearances of the optic nerve head in ADOA are particularly difficult to distinguish from LHON patients with remote visual loss.43 That the morphology of the optic disc in these two conditions should be so strikingly similar is perhaps not surprising in view of some of the presumed common underlying mitochondrial pathophysiological mechanisms.
The importance of a clinical characterisation of the disc in patients with OPA1 ADOA, and its distinction from the disc in glaucoma, is highlighted by the report of the association between two intragenic polymorphisms in the OPA1 gene and normal tension glaucoma (NTG).44 It remains to be seen if the clinical phenotype of NTG patients with these associated polymorphisms is in any way distinct to those NTG patients who do not appear to carry these polymorphisms or from patients with primary open angle glaucoma. However, what is emerging is the possibility that OPA1, and other genes that are now being identified (for example, optineurin OPTN45), may have a role in NTG, possibly as modifier genes.46 Despite considerable similarity, it appears that the disc in OPA1 ADOA is usually distinguishable from that of NTG, although the role of the OPA1 gene in NTG remains to be clarified.
In conclusion, we have described, for the first time, features of the optic disc in patients with ADOA at the OPA1 locus. This may be useful for future studies of phenotype-genotype correlation of the disc in optic neuropathy, including the glaucomas, especially if more genes are found that may influence or modify the glaucoma phenotype. In the long term, continued careful clinical dissection of phenotypes will help to further our understanding of the pathophysiology of atrophy of the optic nerve. The findings of this study suggest that the disc appearance in ADOA differs from NTG chiefly by the shallow shelving. However, there is clearly some overlap. An early age of onset, with a family history and additional clinical features of colour vision deficit, central field involvement with sparing of the peripheral visual field, are important in confirming the diagnosis of ADOA and distinguishing this disc appearance from that of normal tension glaucoma.
Acknowledgments
MV is a Frost Charitable Trust Fellow. This work was supported by the Wellcome Trust, London, and the Guide Dogs for the Blind Association, Berkshire, UK.
REFERENCES
Linked Articles
- Editorial