
Abstract
Keratins are the type I and II intermediate filament proteins which form a cytoskeletal network within all epithelial cells. They are expressed in pairs in a tissue- and differentiation-specific fashion. Epidermolysis bullosa simplex (EBS) was the first human disorder to be associated with keratin mutations. The abnormal keratin filament aggregates observed in basal cell keratinocytes of some EBS patients are composed of keratins K5 and K14. Dominant mutations in the genes encoding these proteins were shown to disrupt the keratin filament cytoskeleton resulting in cells that are less resilient and blister with mild physical trauma.
Identification of mutations in other keratin genes soon followed with attention focussed on disorders showing abnormal clumping of keratin filaments in specific cells. For example, in bullous congenital ichthyosiform erythroderma, clumping of filaments in the suprabasal cells led to the identification of mutations in the suprabasal keratins, K1 and K10. Mutations have now been identified in 18 keratins, all of which produce a fragile cell phenotype. These include ichthyosis bullosa of Siemens (K2e), epidermolytic palmoplantar keratoderma (K1, K9), pachyonychia congenita (K6a, K6b, K16, K17), white sponge nevus (K4, K13), Meesmann’s corneal dystrophy (K3, K12), cryptogenic cirrhosis (K8, K18) and monilethrix (hHb6, hHb1).
In general, these disorders are inherited as autosomal dominant traits and the mutations act in a dominant-negative manner. Therefore, treatment in the form of gene therapy is difficult, as the mutant gene needs to be inactivated. Ways of achieving this are actively being studied. Reliable mutation detection methods from genomic DNA are now available. This enables rapid screening of patients for keratin mutations. For some of the more severe phenotypes, prenatal diagnosis may be requested and this can now be performed from chorionic villus samples at an early stage of the pregnancy.
This review article describes the discovery of, to date, mutations in 18 keratin genes associated with inherited human diseases.
Similar content being viewed by others

1. Keratins
1.1 Structure, Function and Expression
The cytoskeleton of all eukaryotic cells consists of three main networks: microfilaments (5–7nm diameter); microtubules (approximately 25nm diameter); and intermediate filaments (approximately 10nm diameter). Microfilaments and microtubules are involved in various cellular events including cell division, contraction, orientation and polarization, and anchorage. However, the function of intermediate filaments was largely undefined until pathogenic keratin mutations were identified in various skin fragility disorders. This provided increasing evidence that intermediate filaments represent a truly structural cytoskeleton within cells, making them resilient to everyday stress and physical trauma. There are more than 50 intermediate filament proteins, which are classified into at least six types on the basis of sequence homology, tissue specificity and immunologic characteristics. In the cytoplasm of epithelial cells, keratin intermediate filaments form a complex array of filaments from the nucleus to the plasma membrane. At this point they interact with specific protein junctions, desmosomes at intercellular membrane sites and hemidesmosomes at the basal cell surface (figure 1).

(a) Antibody staining of keratin filaments (green) in epithelial cells in culture, nuclei stained with 4c6-diamidino-2-phenylindole⋅2HCI (DAPI) (blue). (b) Abnormal clumping of keratin filaments (green) caused by a mutation in K14 (R125C).
Keratins, of which there are at least 30 cytokeratins and trichocyte keratins (hair/nail keratins), form the type I and II groups of intermediate filament proteins. Analysis of recent sequence from the human genome identified some novel keratins bringing the total number to at least 49.[1] The type I, acidic keratins (K9–K20) are clustered on chromosome 17q12-q21[2,3] and type II, basic keratins (K1–K8) are located in a gene cluster on chromosome 12q11q-14.[2,4] K18 is the exception and maps to the type II gene cluster on chromosome 12.[5] Structurally keratins consist of a central α-helical rod domain of 310 amino acids flanked by non-helical head (V1) and tail (V2) domains (figure 2).[6] The rod domain is divided into the 1A, 1B, 2A and 2B domains each consisting of heptad repeats. Connecting these are three non-helical linkers, L1, L12 and L2 which are thought to provide flexibility to the rod domain. The type II keratins in addition have two subdomains, H1 and H2, which lie between the rod domain and the V1 and V2 domains, respectively. Within the 2B domain there is a ‘stutter’ sequence where the helix polarity is reversed. At the start and end of the rod domain are two short regions: the helix initiation motif at the amino terminus; and the helix termination motif at the carboxy terminus. The sequences of these regions are very highly conserved throughout the intermediate filament protein family. These helix boundary motifs are thought to be critical in overlap interactions during filament assembly.[7] All keratins of a given type have the same genomic organization and position of introns. Type I keratins have 8 exons and 7 introns and type II keratins have 9 exons and 8 introns. The standard nomenclature that is used throughout this review is, for example for keratin 1, K1 for the keratin protein and KRT1 for the corresponding gene.

Schematic diagram showing the basic protein structure of a keratin filament. The α-helical rod domain is divided into four domains, the 1A, 1B, 2A and 2B. Connecting these are the linkers, L1, L12 and L2. Flanking the rod domain are the variable V1 and V2 domains. At either end of the rod domain are the highly conserved helix initiation motif (amino terminus) and helix termination motif (carboxy terminus), shown in red. H1 and H2 subdomains are present in type II keratins. The ‘stutter’ sequence is marked by S.
1.2 Keratin Filament Assembly
The first stage in assembly of a 10nm intermediate filament is the formation of a dimer. Keratins form obligatory heterodimers consisting of a type I and type II keratin. These align in parallel and in axial register to form a coiled coil α-helical dimer. The next stage is the formation of a tetramer by the alignment of two dimers. The precise spatial arrangement of these dimers is still uncertain and has been controversial. Cross-linking experiments indicate that there are three possible ways that keratin dimers could align.[8,9] Tetramers polymerize, both laterally and longitudinally to form 2–3nm protofilaments and when two of these align a 4.5nm protofibril is formed. The inter-twining of protofibrils results in a complete intermediate filament consisting, in the case of keratins, of 32 monomer chains in cross section.[10] The precise arrangement of the keratin filaments to form a three dimensional structure is not yet clear. Recently, some progress has been made in solving the crystal structure of intermediate filament proteins[11] although it is likely to take some years to solve the complete atomic structure of the highly insoluble proteins.
1.3 Keratin Expression Patterns
Keratins are generally expressed in specific pairs, consisting of a type I and a type II keratin in a tissue-specific and differentiation-specific manner[12] as summarized in table I.

Expression patterns of keratins
2. Keratin Diseases
2.1 Epidermolysis Bullosa Simplex (EBS): The First Identified Human Keratin Disease
Epidermolysis bullosa simplex (EBS) was the first inherited disorder to be associated with keratin mutations. To date, mutations in 18 keratin genes are associated with human disorders.
EBS is one of the three main forms of epidermolysis bullosa (EB). The other forms are junctional EB and dystrophic EB. EB classification is on the basis of the ultrastructural level of separation within the skin, an assessment of the clinical features, and the mode of inheritance (autosomal dominant or recessive). The characteristic intra-epidermal blistering of EBS is due to cytolysis in the sub-nuclear region of the basal keratinocytes. EBS is normally inherited as an autosomal dominant trait, although there are rare cases of recessive EBS (see below). In general EBS is considered to be the mildest form of EB with an incidence of approximately 1 : 50 000.[13–15]
The three most common types of autosomal dominant EBS are the Dowling-Meara (section 2.1.1), Weber-Cockayne (section 2.1.2) and Köbner (section 2.1.3) variants, as follows.
2.1.1 EBS Dowling-Meara
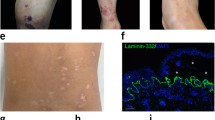
The Dowling-Meara variant (EBS-DM, On-line Mendelian Inheritance in Man [OMIM] 131760) is generally considered to be the most severe form of EBS.[16] Blistering may be extensive at birth and the disorder can be fatal in the first few months, therefore early diagnosis is important to enable appropriate care during this critical period. EBS-DM is inherited in an autosomal dominant manner, although many cases are sporadic. Blisters occur in herpetiform clusters on the trunk and proximal extremities and heal with minor scarring (figure 3) and milia may occur. Oral blistering is common, and the teeth and esophagus may be affected. There is often progressive hyperkeratosis of the palms and soles. Nails may be dystrophic or shed.[17] Histologically, EBS-DM differs from other forms of EBS. Abnormal clumping of keratin filaments within basal keratinocytes is observed. These precede basal cell cytolysis and intraepidermal blister formation.[18–20]

Clinical phenotypes caused by mutations in epidermal keratins. (a) Herpetiform pattern of blisters characteristic of the Dowling-Meara form of epidermolysis bullosa simplex (EBS) [EBS-DM]. Mutations in K5 and K14 underlie EBS. (b) Thickened hyperkeratotic skin typical of bullous congenital ichthyosiform erythroderma (BCIE), is caused by mutations in K1 and K10. (c) Epidermolytic palmoplantar keratoderma (EPPK), hyperkeratosis of the palms showing marked erythematous border is caused by mutations in K9.
2.1.2 EBS Weber-Cockayne
In the Weber-Cockayne form of EBS (EBS-WC, OMIM 131800), blistering predominantly affects the hands and feet.[21,22] Data from the establishment of national EB registries in the UK and in the US suggests that this is the most common form of EBS.[14,15] Blisters develop in early or late childhood, often when a child is learning to walk. The blisters are induced by trauma from routine minor physical activities but can also develop spontaneously. The blisters sometimes bleed but heal without scarring or milia formation. Calluses may develop over areas of recurrent blistering. Hyperhidrosis of the feet is common and there may be mild hyperkeratosis of palms and soles.[23] There is a marked seasonal variation, with more severe blistering occurring in the warmer summer months.
2.1.3 EBS Köbner
EBS Köbner (EBS-K, OMIM 131900) is a subtype similar to EBS-WC but with generalized blistering on all parts of the body and in the oral cavity.[24] However, there is often no clear clinical distinction between EBS-WC and EBS-K as blistering in many patients with EBS-WC is not restricted to the feet and hands. Blisters develop at birth or in early infancy and heal without scarring. Secondary bacterial infections are a common problem. There may be mild to moderate hyperkeratosis of soles and palms and often hyperhidrosis of the feet. The nails may show mild involvement. There is a seasonal variation, like in EBS-WC, with more severe blistering in warm weather.
2.1.4 Identification of Keratin Mutations in EBS
Ultrastructural studies on EBS-DM provided the first indication that an abnormality in a basal cell structural protein could underlie at least some forms of EBS. Abnormal clumping of tonofilaments (keratin filaments) was observed in the basal keratinocytes in association with blistering.[18] Later studies showed tonofilament aggregation in some non-lesional, as well as in peri-lesional skin and in adnexal epithelia including sweat ducts, outer hair root sheaths and sebaceous glands.[19,20] Antibody staining demonstrated that the clumped tonofilaments consisted of the basal cell keratins, K5 and K14. Abnormal round bodies that stained for K5 and K14 were also present in cultured keratinocytes of EBS-DM patients.[19] It was proposed that the filament clumping was a primary event due to specific keratin abnormalities rather than as a consequence of blistering. However, the cause of keratin aggregation and the resulting cell fragility was unknown until studies in several independent laboratories identified the genetic defect in EBS. Using transgenic animals[25,26] and analysis of patient material[27–29] it was predicted and confirmed that mutations in the genes KRT5 and KRT14 that encode keratins K5 and K14 respectively, were the underlying cause of EBS.
Fuchs and colleagues had established the effects of keratin mutants in an in vitro system.[30–32] Expression of keratin mutants in epithelial cells caused aggregation of keratin filaments, (figure 1). To see the effect in vivo, K14 mutations were expressed in the skin of transgenic mice.[25,26] The resultant mice developed intraepidermal blistering at birth or after mild mechanical trauma. A range of EBS-like phenotypes was produced, dependent on the mutation. The most severe phenotype showed aggregation of tonofilaments in skin and in cultured keratinocytes, similar to that observed in EBS-DM.
These findings predicted that different EBS variants could be caused by mutations in K14. DNA sequence analysis was performed on two EBS-DM families. Heterozygous missense mutations were identified in K14, at the same codon arginine 125 (R125) but different bases changes.[28] This arginine is highly conserved throughout evolution. It is located in the helix initiation motif, a region already shown by in vitro deletion experiments to be critical for intermediate filament assembly.
Concurrently, Epstein and colleagues confirmed by genetic linkage analysis and mutation detection that the milder variants, EBS-K and EBS-WC, were also caused by keratin mutations. Linkage of EBS-K in one family to the type I keratin gene cluster on chromosome 17 implicated an abnormality in K14.[33] Consequently, a heterozygous missense mutation in the 2B domain of K14 was identified in this family.[27] In a family affected by EBS-WC, the disorder mapped to the type II keratin gene cluster on chromosome 12. This implied a defect in K5, the expression partner of K14.[33] Additional EBS-K and EBS-WC families were mapped to the keratin gene clusters on chromosomes 12 and 17.[34–36]
A third group simultaneously detected a K5 abnormality in EBS-DM.[29] Keratins expressed by cultured keratinocytes were analyzed by Western blot and stained with monoclonal antibodies. Loss of a K5 epitope on Western blots identified the region of the protein that was mutated. By DNA sequencing, a heterozygous missense mutation was found in the highly conserved helix termination motif of K5.[29]
These first pathogenic keratin mutations in K5 and K14 not only identified the cause of EBS but also confirmed the hypothesis that keratins have a primarily structural role within epithelial cells. They demonstrated that mutations in different members of the heterodimer (K5 or K14) could produce the same phenotype (phenocopies). Also, as predicted by transgenic studies,[25] different EBS phenotypes could be caused by mutations within the same gene.
There are now many reports of mutations in K5 and K14 in EBS-DM and in the milder forms, EBS-K and EBS-WC (keratin mutation database, table II).[37] The majority of these are heterozygous missense mutations. Most mutations in EBS-DM are in the highly conserved helix initiation motif of K14 (table III). Codon R125 has been identified as a ‘hot spot’ for mutations. Mutations at this position occur in a CpG dinucleotide leading to chemical instability, and this may account for the number of mutations found here.[38] Mutations in the conserved helix termination motif of K14 also cause EBS-DM.[39,40] There are fewer mutations in K5 underlying EBS-DM but those reported are also in the helix boundary motifs (keratin mutation database, table II). Some of the more unusual mutations in EBS-DM include a premature stop codon in the helix termination motif of K5,[41] and a donor splice site mutation leading to the deletion of 22 amino acids in the H1 and 1A domains of K5.[42] Another report of a premature stop codon in the helix termination motif of K5 leads to an EBS phenotype with severe blistering in childhood and unusually severe palmoplantar hyperkeratosis.[43] Mutations producing the milder phenotypes EBS-K and EBS-WC have been found throughout the rod domains of K5 and K14 (table III). Surprisingly, some of these are in the L12 linker domain, regions thought to provide some flexibility to the molecule. Mutations in these regions had not been predicted to produce a clinical phenotype. A number of mutations causing EBS-WC have also been found in the H1 head domain of K5. Here, codon 161 is particularly susceptible to mutation (I161S).[44] There is also one report of a deletion mutation in K14 in a case of EBS-WC.[45]

Useful websites for research and diagnosis of keratin disorders

Human keratin gene mutations as at January 2002
Correlations drawn between the positions of mutations versus phenotype suggest the more severe phenotype EBS-DM is caused by mutations in the helix boundary motifs. These domains are predicted to be critical in overlap interactions during filament assembly.[7] Mutations in other regions of the rod domain and just outside in the H1 domain can be better tolerated leading to the milder phenotypes, EBS-K and EBS-WC. However, there are a few exceptions where mutations in the helix termination motif of K14 produce the milder EBS-K and EBS-WC phenotypes.[39] There are also instances where the actual base change together with the position within the rod domain may be important in determining the phenotype. In K14, mutations at codon 119 (M119) can produce all 3 EBS phenotypes. M119T causes EBS-DM, [46,47] M119V causes EBS-K[47] and M119I leads to EBS-WC.[48] Interestingly, one patient from the EBS-WC family was homozygous for the mutation due to a consanguineous marriage and developed a more severe EBS-K phenotype.[49] In this case the mutation is partially dominant unlike the fully recessive ablation mutations (see section 2.1.5) or fully dominant mutations where homozygotes have a similar phenotype to heterozygotes.[50]
2.1.5 Other Forms of EBS with Mutations in K5 and K14
EBS with Mottled Pigmentation
EBS with mottled pigmentation (EBS-MP, OMIM 131960) has been described as a variant of EBS-K.[51] Generalized non-scarring blisters develop at birth or during early infancy. The skin also has a mottled appearance due to a pigmentary abnormality, which fades with age to become barely recognizable in adults. Other associated characteristics include skin atrophy on the trunk and extremities, hyperkeratosis of palms and soles and dystrophic nails. Patients were analyzed for mutations in K5 and K14. The same missense mutation in K5 (L25P) has been identified in all seven cases reported at this time. This mutation is unusual in that it is in the non-conserved variable V1 domain.[52,53] The function of the non-helical V1 and V2 domains at either end of the rod domain are largely unknown, but they are thought to determine the specific function of each keratin. How this mutation produces the pigmentary changes is unknown.
Recessive EBS
In rare instances, EBS can be expressed as a recessive trait (R-EBS, OMIM 601001), in some cases as a result of consanguineous marriages. K14 mutations have been identified in several patients. In the first, a very mild EBS-WC like phenotype was observed in homozygotes. The heterozygous parents were clinically unaffected. The causative mutation was a homozygous missense mutation just outside the helix initiation motif of K14.[54] Several other patients with more severe and generalized blistering have been reported. Complete loss of K14 expression was demonstrated in these patients by immunohistochemistry, immunoelectron microscopy and immunoblotting. K5 expression was normal. These individuals therefore represent human ‘knockouts’ of K14. Homozygous nonsense mutations were detected in two families[55,56] and a homozygous 2 nucleotide deletion mutation in a third family.[57] A homozygous splice site mutation was found in a fourth family.[58] Each mutation resulted in a premature termination codon predicting a truncated K14. Interestingly, a fifth case with a milder phenotype has a homozygous mutation that also results in a premature termination codon. In this case a more truncated protein than in the previous cases is predicted, a K14 protein of only 30 amino acids.[59] A more severe phenotype might have been predicted from this latter mutation but the number of such cases is still too small to draw firm conclusions. In one of the above patients, increased expression of K15 was proposed to compensate for the lack of K14.[58] It appears that the functional relationship between K14 and K15, both type I keratins of basal keratinocytes differs between mice and humans. In contrast to R-EBS patients lacking K14 who can live to old age,[58] transgenic mice in which K14 has been ablated die at about 3 months.[60] In mice, K15 expression decreases with age,[60] whereas in humans, its expression appears to persist and therefore compensates for loss of K14.[58]
As yet there are no cases of recessive EBS with mutations in K5. It is possible that such mutations could be lethal. Evidence for this comes from the recent generation of transgenic mice lacking K5.[61] These mice die at birth due to lack of keratins in the basal cells of the epidermis. Again, this result underlines the ability of K5/K15 filaments to partially rescue the phenotype in neonatal K14 knockout mice, and to a greater degree in R-EBS patients lacking K14. Interestingly, no phenotypes associated with K15 mutations have been reported in either humans or mice, to date.
The identification of keratin mutations in EBS led the way for the molecular basis of other keratin disorders to be elucidated. Disorders where the clinical phenotype affects tissues expressing specific keratins, and that ultrastructurally show aggregation of keratin filaments, were investigated. Mutations have now been identified in 18 keratin genes, either as missense mutations, deletion mutations, insertion mutations, splice site mutations or nonsense mutations. In general, keratin disorders are inherited in an autosomal dominant manner, therefore most keratin mutations act by dominant—negative interference at the protein level. Only a small amount of one copolymerizing keratin need be defective to disrupt the filament network and cause cell fragility. Described below are further keratin diseases for which the molecular pathogenesis has been solved.
2.2 Bullous Congenital Ichthyosiform Erythroderma
Bullous congenital ichthyosiform erythroderma (BCIE), also known as epidermolytic hyperkeratosis (EHK, OMIM 113800), is characterized by erythroderma and blistering at birth, leading to severe generalized hyperkeratosis in adulthood (figure 3). Cytolysis develops in the suprabasal layers of the epidermis, quite distinct from the basal cell blistering in EBS. Ultrastructurally, the basal cells appear normal. However, in the suprabasal cells there is abnormal clumping of tonofilaments leading to a collapsed cytoskeleton in these cells.[62,63] As cells migrate up through the epidermis, K5 and K14 are down regulated and K1 and K10 become the predominant keratins expressed in the suprabasal cells. These keratins became the focus of several studies to determine the molecular basis of BCIE. Again, data from transgenic mice studies supported the idea of a keratin defect. Transgenic mice expressing a truncated human K10 gene gave rise to a phenotype characteristically similar to BCIE.[64] The mutant K10 disrupted normal filament assembly/function in the suprabasal cells producing cytolysis in these upper layers of the epidermis. The first molecular data from patients demonstrated linkage of BCIE to the type II keratin gene cluster on chromosome 12.[65,66] Heterozygous mutations were subsequently identified in several BCIE families in K1 and K10.[67–69] These mutations were in the helix boundary motifs and in the H1 domain of K1. In K10, codon R156, the analogous codon to R125 in K14, was found to be a ‘hot spot’ for mutations.
There have since been numerous reports of mutations in K1 and K10 (table III and keratin mutation database, table II) the majority of them heterozygous missense mutations. In K10, R156 has continued to be the predominant ‘hot spot’ for mutations with 24 of the 38 published K10 mutations occurring here. Mutations in K10 have also been found elsewhere in the helix initiation motif and in the helix termination motif (keratin mutation database, table II). K1 mutations in BCIE are found predominantly in the helix boundary motifs.
From the apparent genotype/phenotype correlation seen in EBS it was predicted that mutations in other regions of the rod domain of K1 or K10 might produce milder BCIE phenotypes. So far there is only one report of such a mutation, a patient with a mild case of BCIE with a mutation in the L12 linker of K1.[70] It may be that some mutations in these regions produce such a mild phenotype that they go undiagnosed. From the mutations reported, BCIE can, however, be divided into two groups. Those with hyperkeratosis of the palms and soles usually have mutations in K1[71] and those without palmoplantar involvement in general are accounted for by mutations in K10.[72] It is postulated that the type I keratin, K9 may be able to partially compensate for disruption to the K10 network. K9 is specifically expressed in palmoplantar skin and this could explain why individuals with mutations in K10 have little or no palmoplantar involvement.[73,74]
Mutations in K1 and K10 have also been found in several patients that did not present with the classical BCIE phenotype; these represent variants of BCIE rather than distinct diseases. One of these is a variant where patients present with epidermal nevi. The areas of skin affected are characterized by a mosaic pattern that follows the lines of Blaschko. Mutations in K1 or K10 have been identified in keratinocytes from lesional skin but not in unaffected skin. These mutations have probably arisen from postzygotic mutations during embryogenesis.[75–77] It is possible that the offspring of these patients may present with generalized BCIE. Individuals with epidermal nevi and with known K1 or K10 mutations should therefore receive appropriate genetic counseling and prenatal diagnosis may be desirable.
Another variant of BCIE described as annular epidermolytic ichthyosis (AEI) is histologically and clinically similar to BCIE.[78] In addition to the blistering, erythema and hyperkeratosis typical of BCIE, numerous annular and polycyclic erythematous hyperkeratotic plaques develop on the trunk and proximal extremities. Mutations in K10 have been found in several patients with AEI.[79,80] More recently K1 mutations have been reported in patients with a similar phenotype, BCIE with cyclic ichthyosis.[81,82]
2.3 Ichthyosis Bullosa of Siemens
Ichthyosis bullosa of Siemens (IBS, OMIM 146800) is a type of epidermolytic hyperkeratosis with epidermal thickening and superficial blistering occurring predominantly on the flexures.[83] Characteristic ‘Mauserung’, or moulting of the outer layers of the epidermis in IBS distinguishes this condition from BCIE. Tonofilament aggregation and cytolysis limited to the upper spinous and granular cell layers of the epidermis was observed in patients with IBS.[84] This made K2e, which is expressed specifically in these upper layers of the epidermis[85] a good candidate gene for IBS. K2e has no obvious expression partner, but is thought to pair with K10. Linkage of the disease to the type II keratin cluster on chromosome 12 supported the idea of KRT2e as a candidate gene.[86] The first of many reports identifying mutations in K2e in IBS followed shortly thereafter.[84,87,88] The mutations were again clustered in the helix boundary motifs. The majority were in the helix termination motif with codon 493, a particular ‘hot spot’ for mutation. In more recent reports this pattern has continued. Of the 26 published cases, 15 patients had codon 493 affected with different substitutions. Of these, 13 were in a CpG dinucleotide sequence involving a G-A transition, E493K (keratin mutation database, table II).
One of the early reports describes K2e mutations in two families that were originally misdiagnosed as BCIE.[88] This is a reminder that there can be overlap in clinical phenotype. Patients with milder forms of BCIE can appear similar clinically to IBS. This should be remembered when determining candidate genes for mutation screening. Using molecular techniques to screen all candidate genes it is possible to confirm the diagnosis in patients that may be difficult to distinguish clinically. It is also well known that for many of these keratin disorders there can be interfamilial phenotypic variation that may make diagnosis more confusing.
2.4 Pachyonychia Congenita (PC)
Pachyonychia congenita (PC) encompasses a range of ectodermal dysplasias where hypertrophic nail dystrophy is a prominent clinical feature. PC is normally inherited in an autosomal dominant manner and is subdivided into two main types:[89] PC-1 (Jadassohn-Lewandowsky type, OMIM 167200)[90] and PC-2 (Jackson-Lawler type, OMIM 167210)[91] Both types are associated with mutations in keratin genes. Mutations in the type II keratin, K6a and its type I expression partner K16 produce PC-1. Mutations in K17 and its recently identified type II expression partner, K6b, produce PC-2.
2.4.1 PC-1
PC-1 is characterized by hypertrophic nail dystrophy, focal non-epidermolytic palmoplantar keratoderma, and oral leukokeratosis (figure 4). Oral leukokeratosis may to a lesser extent be present in PC-2 and is somewhat variable in both forms. By electron microscopy of palmoplantar skin, tonofilaments in suprabasal keratinocytes appear abnormal, as condensed clumps, although these are morphologically different from the aggregates seen in EBS-DM and BCIE.[92,93] These abnormal keratin clumps were proposed to consist of the K6a/K16 expression pair. These keratins are expressed in the nail bed, mucosa and palmoplantar epidermis,[12] the epithelial tissues affected in PC-1. Consequently, K6a and K16 mutation analysis was performed on several PC-1 families. This led to the identification of the first mutations in K6a[94] and K16.[92] Mutations found in additional families are predominantly clustered to the helix initiation motifs (table III). As well as heterozygous missense mutations, several deletion mutations have also been reported in this region. In K6a, codon 171 is a hotspot for either deletion mutation or missense mutation.[95,96]

Clinical phenotypes of pachyonychia congenita (PC) caused by mutations in K6/K16 (PC-1) and K6b/K17 (PC-2). (a) Hypertrophic nail dystrophy typical of PC-1 and PC-2. (b) Oral leukokeratosis, normally present in PC-1, and may to a lesser extent occur in PC-2. (c) Multiple pilosebaceous cysts characteristic of PC-2 patients, occurring post-puberty.
Several reports of mutations in K16 have been found in more unusual PC-1 phenotypes. Firstly, in a case of late onset PC-1 where there were no skin or nail changes until 6 years of age. A missense mutation was found in the central 2B domain of K16.[97] Mutations in this region of K14 cause the milder EBS phenotypes. Secondly, a case described as unilateral palmoplantar verrucous nevus was caused by a mosaic mutation.[98] Although histologically similar to epidermolytic palmoplantar keratoderma (EPPK, see below), the lesions were only present on one side of the body and followed the lines of Blaschko. This is an example of an unusual phenotype where there was no obvious candidate gene. Several candidate genes were screened including K1, K9 and K16. No mutations were found in K1 or K9. However, an in-frame 12 base pair (bp) deletion mutation was identified in the variable V1 domain of K16.[98] This mutation was only detected in cDNA derived from the affected skin of the palm. The normal K16 sequence was found in unaffected skin. Due to the mosaicism observed it was concluded to be a postzygotic mutation.
2.4.2 PC-2
In PC-2, in addition to hypertrophic nail dystrophy, characteristic multiple pilosebaceous cysts develop during puberty (figure 4). These are the main distinguishing features between the two types of PC. Mild focal non-epidermolytic palmoplantar keratoderma, natal teeth, pili torti (twisted hair), and some oral lesions may also be present. In early childhood it can be difficult to distinguish between PC-1 and PC-2. The possibility of a keratin defect underlying PC-2 was explored after linkage analysis of a large Scottish pedigree showed co-segregation of the disease to the type I keratin cluster on chromosome 17.[99] Candidate genes included KRT17, expressed in the outer root sheath of hair follicles, nail bed and sebaceous glands.[100,101] A heterozygous missense mutation in K17 was subsequently found in a PC-2 family.[92] Heterozygous missense mutations in K17 have now been identified in many patients with PC-2 (keratin mutation database, table II). To date, all are in the helix initiation motif, with mutations in codons 92 or 94 accounting for the majority of them. Two deletion mutations, also in the helix initiation motif domain, produce the same PC-2 phenotype as the missense mutations.[96,102] There are no reports of mutations in the helix termination motif of K17 but they are predicted to produce PC-2.
In one family with PC-2, genetic linkage analysis excluded the type I keratin gene cluster, thereby excluding KRT17 as a candidate gene. However, the disease mapped to the type II keratin gene cluster on chromosome 12. There was no obvious type II expression partner for K17 but the K6b isoform of K6 was considered a candidate. It was demonstrated experimentally that K17 and K6b are co-expressed in many epidermal appendages. By mutation analysis, a missense mutation was identified in the helix termination motif of K6b in this family.[103] Thus a second gene for PC-2 was identified.
2.4.3 Steatocystoma Multiplex
Steatocystoma multiplex (OMIM 184500), a variant of PC-2, is characterized by multiple pilosebaceous cysts like those seen in PC-2. There are no additional features other than perhaps very mild nail involvement. Missense mutations in K17 have been found in three families.[104,105] Interestingly, the mutations reported occur at the same codons as those that can also produce PC-2. In one case the mutation was the same, R94C.[104]
As seen for other keratins phenotypic variation is also seen with mutations in K17. Different phenotypes (PC-2 or steatocystoma multiplex) can arise from the same mutation, indicating that the phenotype must depend on a combination of other genetic and environmental factors.
2.5 Palmoplantar Keratoderma
The inherited palmoplantar keratodermas encompass a range of disorders that can be difficult to distinguish clinically. Hyperkeratosis of the palms and soles occurs in a diffuse, focal or punctate pattern and may be associated with other ectodermal features.[106] Different forms of palmoplantar keratoderma are caused by mutations in different keratin genes, for example epidermolytic palmoplantar keratoderma is caused by mutations in K9 (see section 2.5.1). Mutations in other forms of palmoplantar keratoderma have been found in keratin genes that are principally involved in more common diseases. For example, focal non-epidermolytic palmoplantar keratoderma is caused by mutations in K16 (see below), mutations in K16 also cause PC-1, as described above. The majority of mutations in K1 result in BCIE but a few unusual cases of palmoplantar keratoderma have been reported that also have K1 mutations. These findings demonstrate the heterogeneous phenotype that can result from mutations in some keratin genes.
2.5.1 Epidermolytic Palmoplantar Keratoderma (EPPK)
Characterized by epidermolytic hyperkeratosis of palmoplantar epidermis, epidermolytic palmoplantar keratoderma (EPPK, OMIM 144200) is an autosomal dominant disorder that presents early in life.[107] Histologically, cytolysis occurs in the suprabasal layers of thick epidermis and aggregates of keratin filaments are seen by electron microscopy.[108] The distinct localization of the diffuse thick yellowish hyperkeratosis to the palms and soles with no other clinical phenotype (figure 3) pointed towards KRT9 as the candidate gene because of its specific expression pattern in the suprabasal cells of palmoplantar skin.[73,74] Like K2e, K9 appears to be an accessory keratin probably pairing with K1 but with no other obvious type II keratin expression partner. The first EPPK families studied mapped to the type I keratin cluster on chromosome 17.[109] Following the cloning of the K9 cDNA and corresponding gene, mutation analysis was performed in patients with EPPK.[110] Heterozygous missense mutations were identified in the helix initiation motif of K9 in several EPPK families.[110] To date, 28 of the 29 mutations reported for EPPK are in the helix initiation motif (table III), 16 of these are in codon 162 (R162) the analogous codon to R125 in K14 and R156 in K10. These are CpG ‘hot spot’ mutations in EBS and BCIE respectively. Similarly, mutations in K9 R162 occur in a CpG dinucleotide. The only K9 mutation reported that does not occur in the 1A domain is a 3 bp insertion mutation in the helix termination motif.[111] This was the first report of an insertion mutation in a human keratin and the clinical phenotype was shown to be similar to those caused by missense mutations.
2.5.2 Mild EPPK
In three families with a mild form of EPPK, linkage of the disease to the type II keratin gene cluster eliminated KRT9 as a candidate gene. Analysis of KRT1 identified a splice site mutation at the exon 6 splice donor site. This resulted in a new splice site 54 bases downstream and subsequent insertion of 18 bp.[112] Genotyping data suggested that all three families could have the same ancestral mutation. The disease phenotype in these families is quite distinct from BCIE.
2.5.3 Diffuse Non-Epidermolytic Palmoplantar Keratoderma
An unusual mutation has been reported in the Unna-Thost form of PPK. Linkage analysis excluded the type I keratin locus and therefore KRT9 was excluded as a candidate gene.[113] KRT1 was screened and a missense mutation was found. But unlike all other K1 mutations this is in the non-conserved variable V1 domain at the amino terminus. No further mutations have been reported in non-epidermolytic palmoplantar keratoderma (NEPPK).
2.5.4 Ichthyosis Hystrix Curth-Macklin
A case of severe mutilating palmoplantar keratoderma, present through several generations in an African-American family, was recently reported as the Curth-Macklin form of ichthyosis hystrix (IHCM, OMIM 146590). Histologically, the keratin filaments in the spinous and granular layer appeared abnormal. Linkage analysis of several candidate regions showed complete co-segregation of the disease with the type II keratin cluster on chromosome 12.[114] Several type II keratin genes were screened for mutations. A mutation was identified in K1 in the non-conserved variable V2 domain at the carboxy terminus. Substitution of two guanine bases for an adenine resulted in a frameshift and premature termination codon 229 bp downstream.[114] This is the first pathogenic mutation occurring in the V2 domain of a keratin. Interestingly, the keratin loci were eliminated in another family diagnosed as IHCM.[115]
2.5.5 Focal Non-Epidermolytic Palmoplantar Keratoderma
Focal non-epidermolytic palmoplantar keratoderma (FNEPPK) is characterized by the presence of focal hyperkeratosis on the pressure points of the palms and soles similar to that observed in PC-1. There is minimal or no nail involvement and no oral involvement. In two individuals presenting with FNEPPK, heterozygous missense mutations were found in the helix initiation motif of K16.[116] These mutations are similar to those causing PC-1 and might have been predicted to give a more severe phenotype. This again demonstrates the complexity of keratin mutations. A third case is an unusual heterozygous mutation, a 23 bp deletion and a separate 1 bp deletion in the helix termination motif.[117] This removes most of the helix termination motif, the most highly conserved motif sequence in keratins and all intermediate filaments. The unexpectedly mild phenotype suggests that loss of most of this critical domain has a milder dominant-negative effect on filament assembly than certain missense mutations. Data from transient transfections of mutant K16 protein and other K16 constructs into epithelial cells in culture supported this hypothesis.[117] Mutations in K6a are also predicted to give a FNEPPK phenotype.
2.6 Non-Epidermal Keratin Disorders
Certain keratins are expressed in tissues other than the epidermis and epidermal appendages. Mutations in these keratins were also proposed to result in a clinical phenotype. The first mutations to be identified in a non-epidermal keratin were in the disorder white sponge nevus.
2.6.1 White Sponge Nevus
White sponge nevus of Canon (WSN, OMIM 193900) occurs as a benign autosomal dominant disorder affecting non-cornifying stratified squamous epithelia. White ‘spongy’ plaques are observed, predominantly in the mouth (oral leukokeratosis) (figure 5) but may also be present in the esophagus and anogenital mucosa.[118] Onset is often in early childhood. Reports of tonofilament aggregation in patients with WSN indicated a defect in K4 or K13.[119,120] These keratins are specifically expressed in the mucosal non-cornifying stratified epithelia. Mutation analysis of two Scottish families revealed a 3 bp heterozygous deletion mutation in the helix initiation motif of K4.[121] Independently, another group identified a heterozygous missense mutation in K13 in a family with WSN.[122] Further heterozygous mutations have been identified in other WSN families including a 3 bp insertion in K4[123] and missense mutations in K13.[124,125] To date, the mutations in K4 and K13 have all been found in the helix initiation motif (table III) but one would predict that mutations in the helix termination motif would produce a similar phenotype.

(a) Oral white sponge nevus (WSN), oral leukokeratosis is due to mutations in K4 and K13 (reproduced from Munro,[126] with permission from Dermatology in Practice). (b) Meesmaan’s epithelial corneal dystrophy (MECD), multiple fine microcysts in the cornea are seen by slit lamp due to mutations in K3 and K12.
2.6.2 Meesmann’s Epithelial Corneal Dystrophy
Meesmann’s epithelial corneal dystrophy (MECD, OMIM 122100) is an autosomal dominant disorder of the corneal epithelium originally described in a large German family.[127] There are characteristic intraepithelial cysts in the anterior cornea seen by slit lamp (figure 5). These cysts are filled with intracellular debris and probable keratin aggregates.[128] Onset is in early childhood although it may sometimes be delayed until late childhood/early adulthood. The disorder is often asymptomatic, and vision is not normally affected. However, because of the fragility of the corneal epithelium, patients can be intolerant of contact lenses. K3 and K12 are specifically expressed in the corneal epithelial cells.[12,129] This, together with the fact that K12 knockout mice have extremely fragile corneal keratinocytes,[130] led to the discovery that mutations in K3 and K12 can cause this disorder.[131] Descendants of Meesmann’s original German kindred were studied and by genetic linkage analysis MECD was mapped to the KRT12 locus. Co-segregation of this locus was also found in a small family from Northern Ireland. Mutation analysis identified heterozygous missense mutations in the helix initiation motif of K12 in both families. In a second family from Northern Ireland, the disorder co-segregated with the KRT3 locus and consequently a mutation was identified in helix termination motif of K3.[131] Mutations have been identified in other families, all occurring in the helix boundary motifs of K12.[132–135]
2.6.3 Cryptogenic Cirrhosis
The simple epithelial keratins K8 and K18 are expressed in the liver, pancreas and intestinal epithelium. For some time, keratin researchers wondered what clinical phenotype would be produced by mutations in these genes. Good evidence for association of these genes and liver disorders came from the discovery that transgenic mice expressing mutant K18 develop chronic hepatitis.[136] DNA was then analyzed from patients with liver disease of unknown cause. In one out of 28 patients with cryptogenic cirrhosis, a missense mutation was identified in K18 (H127L).[137] This mutation is unusual in that it is in the L1 linker domain, a site where no other pathogenic keratin mutations have been reported. A follow up study of a large number of patients from three groups: patients with cryptogenic liver disease, noncryptogenic liver disease, and randomly selected patients who provided blood to the hematology department, were analyzed for mutations in K8 or K18. Five mutations were identified in the H1 domain of K8 in patients with cryptogenic liver disease, three were G61C and two were Y53H.[138] Mutations in the H1 domain of other type II keratins have been shown to be pathogenic. In K1 and K5, mutations in the H1 domain underlie some cases of BCIE and EBS-WC respectively (keratin mutation database, table II). The effect of these K8 mutations on filament reorganization were analyzed in tissue culture experiments.[138] Together with data from transgenic studies, the results indicate that mutations in K8 or K18 may predispose patients to late onset liver disease.
2.6.4 Trichocyte (Hair) Keratins
The type I and type II trichocyte or hair keratin genes co-localize with the cytokeratins on chromosome 17 and 12 respectively. These keratins form the highly cross-linked tough polymer of which hair, nail and the hard tips of the lingual papillae are composed. These proteins share the same basic structure as epithelial keratins but are very rich in cysteine residues, allowing a high degree of disulfide cross-linking. The proposal that mutations in these genes could produce an abnormal hair phenotype has been investigated.
Monilethrix
One candidate disease for mutations in a trichocyte keratin was monilethrix (OMIM 158000). This is a rare autosomal dominant disease with variable phenotypic expression, from mild hair loss to near total alopecia (figure 6). The hair is fragile and by light microscopy, periodic thinning of the hair shaft gives the hair a beaded appearance. By electron microscopy, defects are found in the microfibrillar structure of the hair shaft. Cytolysis and tonofilament clumping in the cortical cells of the bulb of the hair follicle are also observed.[139] Follicular keratosis and nail abnormalities may be present. Evidence for a defect in a trichocyte keratin came from genetic linkage between monilethrix and the type II keratin gene cluster on chromosome 12.[140,141] Identification of heterozygous missense mutations in the type II hair keratin hHb6 followed.[142] In all patients, the mutation occurred in the helix termination motif in codon 413 but by different substitutions. A subsequent report identified further mutations in hHb6, again in codon 413 thereby identifying it as a ‘hot spot’ for mutations.[143] Additionally, a heterozygous missense mutation was identified in another family in the corresponding residue of the highly related type II keratin, hHb1.[143] Mutations have now been identified in many families (keratin mutation database, table II). To date, all are missense mutations occurring predominantly in the helix termination motif of hHb6 (table III). A few have been reported in the helix initiation motif of hHb6, and several in the helix termination motif of hHb1 (table III).

Monilethrix, variable alopecia, caused by mutations in hair keratins hHb6 or Hhb1.
3. Molecular Genetics in Diagnosis of Keratin Disorders
Before identification of keratin mutations in the disorders described above, diagnosis was often confirmed by histological analysis of a skin biopsy. For example, to determine the level of splitting within the blister in EBS, or to look for the characteristic clumping of keratin filaments in EBS-DM. These histological methods are important and are sometimes still necessary. However, by using molecular techniques the exact gene defect can be identified, providing an unequivocal diagnosis. These can be performed on genomic DNA derived from blood samples, alleviating the need to take biopsy material. Analysis at the molecular level is by DNA sequencing of candidate genes. In patients not fitting the phenotype classification, genetic linkage analysis is performed to determine the keratin gene cluster to which the disease maps. Candidate genes are then screened by DNA sequencing. Mutations should be present in all affected family members but absent in unaffected members. Confirmation of a mutation also requires screening of at least 50 ethnically matched, unrelated, control DNA samples to exclude the mutation as a common polymorphism occurring within the general population. For all keratins associated with diseases at this time (except K8 and K18) there are two ‘hot spot’ regions for mutations, the highly conserved helix boundary motifs. Mutation screening should focus on these regions first. In addition, the H1 domain of type II keratins and the L12 linker domain of both types are frequent sites for mutations.
In rare cases where inheritance of the disease is recessive, staining of tissue from patients with appropriate antibodies may reveal loss of expression of a particular keratin. DNA sequencing of this candidate gene will identify the mutation. In consanguineous families, linkage analysis by homozygosity mapping may be useful.
3.1 Pseudogenes
The presence of multiple pseudogenes for some keratins, some of which are also expressed at low levels, hampered early mutation detection studies using genomic DNA samples. It is important to be aware of pseudogenes to ensure that mutation analysis is only performed on the functional gene. Pseudogenes have been reported for K6,[144] K8,[1] K14,[145] K16,[3] K17,[146] K18,[1] and K19.[147] The sequences of some of these pseudogenes are now known. It is therefore possible to design specific polymerase chain reaction (PCR) primers for the respective functional gene to avoid amplification of pseudogenes.
There is at least one KRT14 pseudogene.[145] A genomic mutation detection system has been developed for exons 1, 4 and 6 which encode the 1A, L12 and 2B domains of K14 containing the mutation ‘hot spots’.[39] In this case, before PCR amplification, genomic DNA is digested with restriction enzymes that cleave only the KRT14 pseudogene, leaving the genomic sequences of the functional KRT14 intact.
Several discrepancies in the published K16 coding sequence.[3,148,149] led to a further study.[150] This confirmed the K16 cDNA sequence reported by Paladini et al.[149] and described cloning of the corresponding KRT16 gene and 2 pseudogenes. PCR primers were designed to specifically amplify the functional KRT16.[150] Mutations identified in KRT16 in PC-1 families were also confirmed by reverse transcription (RT)-PCR in mRNA from the same individuals demonstrating specificity of genomic PCR for the functional gene
Likewise for KRT17, specific PCR conditions have been developed[92,96] to avoid amplification of the two reported KRT17 pseudogenes.[146]
For K6 there are at least six copies of the gene, of which at least two are expressed.[144] A mutation detection strategy has been developed for all of KRT6a,[151] and some exons of KRT6b.[103] Several mutations identified in genomic DNA from PC patients have been confirmed at the mRNA level, demonstrating specificity of these specific PCR primers and conditions.
4. Future Studies
As more patients are studied, additional mutations may be found outside the hotspot regions. This should provide further information regarding the function of these domains. In some cases there may be no mutation in the candidate keratin genes. In these situations it is worth considering the few known keratins that are not yet associated with a human disorder, for example, K15. It is also probable that some of the novel keratin genes identified from sequence data from the human genome project may be associated with human diseases.[1] Alternatively mutations in other genes such as those encoding intermediate filament associated proteins may produce similar phenotypes to those of some keratin genes. For example, a form of EBS with muscular dystrophy is due to mutations in plectin, a high molecular weight cytoskeleton associated protein.[152,153] Mutations in the desmosomal protein, plakophilin 1, lead to an ectodermal dysplasia phenotype with skin fragility.[154]
5. Prenatal Diagnosis
Now that many keratin disorders can be diagnosed at the molecular level, the focus of several research groups is on treatment and prevention of these diseases. Treatment in the form of gene therapy is difficult as most keratin mutations are dominant acting and the mutant gene needs to be inactivated. Ways of achieving this are currently being studied. A mouse model has recently been developed that provides a realistic model of EBS-DM. This has important implications for gene therapy of EBS and will provide a system to test different approaches in vivo.[155]
While many of the disorders caused by keratin mutations are not severe enough to warrant prenatal diagnosis, in some of the more severe phenotypes such as EBS-DM, BCIE or severe PC, it may be requested. Previously diagnosis would have been from a fetal skin biopsy, for example in the case of EBS, to determine the presence of blistering. However, reliable mutation detection methods from genomic DNA are now available. Therefore, it is possible when the familial mutation is known, to perform prenatal diagnosis from chorionic villus samples (CVS) at an early stage of the pregnancy. CVS DNA is obtained at 11–12 weeks gestation for mutation analysis. Multiple villi are dissected out from the CVS and DNA is isolated using standard DNA extraction protocols. Care is needed to prevent contamination of the CVS DNA with maternal cells as this can lead to problems of diagnosis. CVS DNA is screened for the known familial keratin mutation using standard PCR and DNA sequencing protocols. Prenatal diagnosis by mutation analysis has been performed for several patients with BCIE,[156,157] EBS,[40] and one patient with PC-1.[150] In the UK, genetic diagnosis is performed by National Health Service laboratories, and in the USA by local genetics laboratories or by private companies (see table II).
6. Intermediate Filament Mutation Database
With the increasing number of mutations identified within the keratins, together with the pathogenic mutations that are emerging in other members of the intermediate filament gene superfamily, there was a need for a readily accessible database of this information. The Epithelial Genetics group at Dundee University, at the suggestion of the HUGO Human Genome Variation Society, has established the Intermediate Filament Mutation Database to address this problem. This catalog of mutations will provide a valuable resource when analyzing mutations in terms of their frequency, position and clinical phenotype, and allow correlations to be made between genotype and phenotype. A summary of the number of keratin mutations as at January 2002 is shown in table III. This database is regarded as the property of all keratin and intermediate filament researchers and will be run as an international consortium. Comments and suggestions are welcome, as well as direct submission of mutations and polymorphisms, at the website (http://www.interfil.org), see table II.
7. Conclusion
During the last decade mutations in 18 keratin genes have been associated with epithelial diseases. This has led to improved classification of these disorders and diagnosis by molecular techniques combined with clinical examination and histological analysis. Current research in many groups is now focussed on developing treatments to alleviate the debilitating symptoms caused by these diseases.
References
Hesse M, Magin TM, Weber K. Genes for intermediate filament proteins and the draft sequence of the human genome: novel keratin genes and a surprisingly high number of pseudogenes related to keratin genes 8 and 18. J Cell Sci 2001; 114 (Pt 14): 2569–2575
Romano V, Bosco P, Rocchi M, et al. Chromosomal assignments of human type I and type II cytokeratin genes to different chromosomes. Cytogenet Cell Genet 1988; 48: 148–151
Rosenberg M, RayChaudhury A, Shows T, et al. A group of type I keratin genes on human chromosome 17: characterization and expression. Mol Cell Biol 1988; 8: 722–736
Rosenberg M, Fuchs E, LeBeau MM, et al. Three epidermal and one simple epithelial type II keratin genes map to human chromosome 12. Cytogenet Cell Genet 1991; 57: 33–38
Waseem A, Alexander CM, Steel JB, et al. Embryonic simple epithelial keratins 8 and 18: chromosomal location emphasizes difference from other keratin pairs. New Biol 1990; 2 (5): 464–478
Smith T A, Strelov SV, Burkhard P, et al. Sequence comparisons of intermediate filament chains: evidence of a unique functional/structural role for coiled-coil segment 1A and linker L1. J Struct Biol 2002; 137: 128–145
Steinert PM, Yang JM, Bale SJ, et al. Concurrence between the molecular overlap regions in keratin intermediate filaments and the locations of keratin mutations in genodermatoses. Biochem Biophys Res Commun 1993; 197 (2): 840–848
Steinert PM. Structure, function and dynamics of keratin intermediate filaments. J Invest Dermatol 1993; 100 (6): 729–734
Steinert PM, North AC, Parry DA. Structural features of keratin intermediate filaments. J Invest Dermatol 1994; 103 (5 Suppl.): 19S–24S
Steven AC, Hainfeld JF, Trus BL, et al. Epidermal keratin filaments assembled in vitro have masses per unit length that scale according to average subunit mass: structural basis for homologous packing of subunits in intermediate filaments. J Cell Biol 1983; 97: 1939–1944
Herrmann H, Strelkov SV, Feja B, et al. The intermediate filament protein consensus motif of helix 2B: its atomic structure and contribution to assembly. J Mol Biol 2000; 298 (5): 817–832
Moll R, Franke WW, Schiller DL, et al. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell 1982; 31 (1): 11–24
McKenna KE, Walsh MY, Bingham EA. Epidermolysis bullosa in Northern Ireland. Br J Dermatol 1992; 1277 (4): 318–321
Fine JD. Epidermolysis bullosa: application of epidemiologic principles to the study of a group of rare diseases via a disease registry. Dermatol Clin 1995; 13 (3): 659–670
Horn HM, Priestley GC, Eady RA, et al. The prevalence of epidermolysis bullosa in Scotland. Br J Dermatol 1997; 136 (4): 560–564
Dowling GB, Meara RH. Epidermolysis bullosa resembling juvenile dermatitis herpetiformis. Br J Dermatol 1954; 66: 139–143
Fine JD, Bauer EA, Briggaman RA, et al. Revised clinical and laboratory criteria for subtypes of inherited epidermolysis bullosa: a consensus report by the subcommittee on diagnosis and classification of the national epidermolysis bullosa registry. J Am Acad Dermatol 1991; 24: 119–135
Anton-Lamprecht I, Schnyder UW. Epidermolysis bullosa herpetiformis Dowling-Meara: report of a case and pathomorphogenesis. Dermatologica 1982; 164: 221–235
Ishida-Yamamoto A, McGrath JA, Chapman SJ, et al. Epidermolysis bullosa simplex (Dowling-Meara type) is a genetic disease characterized by an abnormal keratin filament network involving keratins K5 and K14. J Invest Dermatol 1991; 97 (6): 959–968
McGrath JA, Ishida-Yamamoto A, Tidman MJ, et al. Epidermolysis bullosa simplex (Dowling-Meara): a clinicopathological review. Br J Dermatol 1992; 126: 421–430
Weber F. Recurrent bullous eruptions on the feet in a child. Froc R Soc Med 1926; 19: 72
Cockayne E. Recurrent bullous eruptions of the feet. Br J Dermatol 1938; 55: 358–362
Pearson RW. Clinicopathologic types of epidermolysis bullosa and their nondermatological complications. Arch Dermatol 1988; 124 (5): 718–725
Koebner H. Hereditaere Anlage zur Blasenbildung (Epidermolysis bullosa hereditaria). Dtsch Med Wochenschr 1886; 12: 21–22
Coulombe P A, Hutton ME, Vassar R, et al. A function for keratins and a common thread among different types of epidermolysis bullosa simplex diseases. J Cell Biol 1991; 115: 1661–1674
Vassar R, Coulombe P A, Degenstein L, et al. Mutant keratin expression in transgenic mice causes marked abnormalities resembling a human genetic skin disease. Cell 1991; 64: 365–380
Bonifas JM, Rothman AL, Epstein Jr EH. Epidermolysis bullosa simplex: evidence in two families for keratin gene abnormalities. Science 1991; 254 (5035): 1202–1205
Coulombe PA, Hutton ME, Letai A, et al. Point mutations in human keratin 14 genes of epidermolysis bullosa simplex patients: genetic and functional analysis. Cell 1991; 66: 1301–1311
Lane EB, Rugg EL, Navsaria H, et al. A mutation in the conserved helix termination peptide of keratin 5 in hereditary skin blistering. Nature 1992; 356 (6366): 244–246
Albers K, Fuchs E. The expression of mutant epidermal keratin cDNAs transfected in simple epithelial and squamous cell carcinoma lines. J Cell Biol 1987; 105: 791–806
Albers K, Fuchs E. Expression of mutant keratin cDNAs in epithelial cells reveals possible mechanisms for initiation and assembly of intermediate filaments. J Cell Biol 1989; 108: 1477–1493
Coulombe P A, Chan YM, Albers K, et al. Deletions in epidermal keratins leading to alterations in filament organization in vivo and in intermediate filament assembly in vitro. J Cell Biol 1990; III (6 Pt 2): 3049–3064
Bonifas JM, Rothman AL, Epstein E. Linkage of epidermolysis bullosa simplex to probes in the region of keratin gene clusters on chromosomes 12q and 17q [abstract]. J Invest Dermatol 1991; 96: 550a
Ryynanen M, Knowlton RG, Uitto J. Mapping of epidermolysis-bullosa simplex mutation to chromosome-12. Am J Hum Genet 1991; 49 (5): 978–984
Hoyheim B, Gedde-Dahl T, Olaisen B. Linkage studies in epidermolysis bullosa simplex [abstract]. J Invest Dermatol 1992; 98: 397a
McKenna KE, Hughes AE, Nevin NC. Linkage of epidermolysis bullosa simplex to keratin gene loci. J Med Genet 1992; 29: 568–570
Corden LD, McLean WH. Human keratin diseases: hereditary fragility of specific epithelial tissues. Exp Dermatol 1996; 5 (6): 297–307
Cooper DN, Krawczak M. Human gene mutation. Oxford: BIOS Scientific Publishers Ltd, 1993
Hut PH, v d Vlies P, Jonkman MF, et al. Exempting homologous pseudogene sequences from polymerase chain reaction amplification allows genomic keratin 14 hotspot mutation analysis. J Invest Dermatol 2000; 114 (4): 616–619
Rugg EL, Baty D, Shemanko CS, et al. DNA based prenatal testing for the skin blistering disorder epidermolysis bullosa simplex. Prenat Diagn 2000; 20 (5): 371–377
Muller FE, Anton-Lamprecht I, Kuster W, et al. A premature stop codon mutation in the 2B helix termination peptide of keratin 5 in a German epidermolysis bullosa simplex Dowling-Meara case. J Invest Dermatol 1999; 112 (6): 988–990
Rugg EL, Rachet-Prehu MO, Rochat A, et al. Donor splice site mutation in keratin 5 causes in-frame removal of 22 amino acids of H1 and 1A rod domains in Dowling-Meara epidermolysis bullosa simplex. Eur J Hum Genet 1999; 7 (3): 293–300
Livingston RJ, Sybert VP, Smith LT, et al. Expression of a truncated keratin 5 may contribute to severe palmar: plantar hyperkeratosis in epidermolysis bullosa simplex patients. J Invest Dermatol 2001; 116 (6): 970–974
Ehrlich P, Sybert VP, Spencer A, et al. A common keratin 5 gene mutation in epidermolysis bullosa simplex Weber-Cockayne. J Invest Dermatol 1995; 104 (5): 877–879
Chen MA, Bonifas JM, Matsumura K, et al. A novel three-nucleotide deletion in the helix 2B region of keratin 14 in epidermolysis bullosa simplex: DE375. Hum Molec Genet 1993; 2 (11): 1971–1972
Shemanko CS, Mellerio JE, Tidman MJ, et al. Severe palmo-plantar hyperkeratosis in Dowling-Meara epidermolysis bullosa simplex caused by a mutation in the keratin 14 gene (KRT14). J Invest Dermatol 1998; 111 (5): 893–895
Cummins RE, Klingberg S, Wesley J, et al. Keratin 14 point mutations at codon 119 of helix 1A resulting in different epidermolysis bullosa simplex phenotypes. J Invest Dermatol 2001; 117 (5): 1103–1107
Chen H, Bonifas JM, Matsumura K, et al. Keratin 14 gene mutations in patients with epidermolysis bullosa simplex. J Invest Dermatol 1995; 105 (4): 629–632
Hu ZL, Smith L, Martins S, et al. Partial dominance of a keratin 14 mutation in epidermolysis bullosa simplex: increased severity of disease in a homozygote. J Invest Dermatol 1997; 109 (3): 360–364
Stephens K, Zlotogorski A, Smith L, et al. Epidermolysis bullosa simplex: a keratin 5 mutation is a fully dominant allele in epidermal cytoskeleton function. Am J Hum Genet 1995; 56 (3): 577–585
Fischer T, Gedde-Dahl Jr T. Epidermolysis bullosa simplex and mottled pigmentation: a new dominant syndrome: 1. clinical and histological features. Clin Genet 1979; 15 (3): 228–238
Uttam J, Hutton E, Coulombe PA, et al. The genetic basis of epidermolysis bullosa simplex with mottled pigmentation. Proc Natl Acad Sci USA 1996; 93 (17): 9079–9084
Irvine AD, Rugg EL, Lane EB, et al. Molecular confirmation of the unique phenotype of epidermolysis bullosa simplex with mottled pigmentation. Br J Dermatol 2001; 144 (1): 40–45
Hovnanian A, Pollack E, Hilal L, et al. A missense mutation in the rod domain of keratin 14 associated with recessive epidermolysis bullosa simplex. Nat Genet 1993; 3: 327–332
Chan Y, Anton-Lamprecht I, Yu QC, et al. A human keratin 14 “knockout”: the absence of K14 leads to severe epidermolysis bullosa simplex and a function for an intermediate filament protein. Genes Dev 1994; 8 (21): 2574–2587
Corden LD, Mellerio JE, Gratian MJ, et al. Homozygous nonsense mutation in helix 2 of K14 causes severe recessive epidermolysis bullosa simplex. Hum Mutat 1998; 11: 279–285
Rugg EL, McLean WHI, Lane EB, et al. A functional “knock-out” for human keratin 14. Genes Dev 1994; 8 (21): 2563–2573
Jonkman MF, Heeres K, Pas HH, et al. Effects of keratin 14 ablation on the clinical and cellular phenotype in a kindred with recessive epidermolysis bullosa simplex. J Invest Dermatol 1996; 107: 764–769
Batta K, Rugg EL, Wilson NJ, et al. A keratin 14 ‘knockout’ mutation in recessive epidermolysis bullosa simplex resulting in less severe disease. Br J Dermatol 2000; 143 (3): 621–627
Lloyd C, Yu QC, Cheng J, et al. The basal keratin network of stratified squamous epithelia: defining K15 function in the absence of K14. J. Cell Biol 1995; 129 (5): 1329–1344
Peters B, Kirfel J, Bussow H, et al. Complete cytolysis and neonatal lethality in keratin 5 knockout mice reveal its fundamental role in skin integrity and in epidermolysis bullosa simplex. Mol Biol Cell 2001; 12 (6): 1775–1789
Anton-Lamprecht I. Genetically induced abnormalities of epidermal differentiation and ultrastructure in ichthyosis and epidermolysis: pathogenesis, heterogeneity, fetal manifestations, and prenatal diagnosis. J Invest Dermatol 1983; 81 (1 Suppl.): 149s–156s
Ishida-Yamamoto A, McGrath JA, Judge MR, et al. Selective involvement of keratins K1 and K10 in the cytoskeletal abnormality of epidermolytic hyperkeratosis (bullous congenital ichthyosiform erythroderma). J Invest Dermatol 1992; 99: 19–26
Fuchs E, Esteves RA, Coulombe PA. Transgenic mice expressing a mutant K10 gene reveal the likely genetic basis for epidermolytic hyperkeratosis. Proc Natl Acad Sci USA 1992; 89: 6906–6910
Bonifas JM, Bare JW, Chen MA, et al. Linkage of the epidermolytic hyperkeratosis phenotype and the region of the type II keratin gene cluster on chromosome 12. J Invest Dermatol 1992; 99: 524–527
Compton JG, DiGiovanna JJ, Santucci SK, et al. Linkage of epidermolytic hyperkeratosis to the type II keratin gene cluster on chromosome 12q. Nature Genet 1992; 1: 301–305
Cheng J, Syder AJ, Yu Q-C, et al. The genetic basis of epidermolytic hyperkeratosis: a disorder of differentiation-specific epidermal keratin genes. Cell 1992; 70: 811–819
Chipev CC, Korge BP, Markova N, et al. A leucine-proline mutation in the H1 subdomain of keratin 1 causes epidermolytic hyperkeratosis. Cell 1992; 70: 821–828
Rothnagel JA, Dominey AM, Dempsey LD, et al. Mutations in the rod domains of keratins 1 and 10 in epidermolytic hyperkeratosis. Science 1992; 257: 1128–1130
Kremer H, Lavrijsen AP, McLean WH, et al. An atypical form of bullous congenital ichthyosiform erythroderma is caused by a mutation in the L12 linker region of keratin 1. J Invest Dermatol 1998; 111 (6): 1224–1226
DiGiovanna JJ, Bale SJ. Epidermolytic hyperkeratosis: applied molecular genetics. J Invest Dermatol 1994; 102 (3): 390–394
Yang JM, Nam K, Kim SW, et al. Arginine in the beginning of the 1A rod domain of the keratin 10 gene is the hot spot for the mutation in epidermolytic hyperkeratosis. J Dermatol Sci 1999; 19 (2): 126–133
Knapp AC, Franke WW, Heid H, et al. Cytokeratin no. 9, an epidermal type I keratin characteristic of a special program of keratinocyte differentiation displaying body site specificity. J Cell Biol 1986; 103: 657–667
MoH I, Heid H, Franke WW, et al. Distribution of a special subset of keratinocytes characterized by the expression of cytokeratin 9 in adult and fetal human epidermis of various body sites. Differentiation 1987; 33: 254–265
Paller AS, Syder AJ, Chan YM, et al. Genetic and clinical mosaicism in a type of epidermal nevus. N Engl J Med 1994; 331 (21): 1408–1415
Moss C, Jones DO, Blight A, et al. Birthmark due to cutaneous mosaicism for keratin 10 mutation [letter]. Lancet 1995; 345 (8949): 596
Nomura K, Umeki K, Hatayama I, et al. Phenotypic heterogeneity in bullous congenital ichthyosiform erythroderma: possible somatic mosaicism for keratin gene mutation in the mildly affected mother of the proband. Arch Dermatol 2001; 137 (9): 1192–1195
Sahn EE, Weimer Jr CE, Garen PD. Annular epidermolytic ichthyosis: a unique phenotype. J Am Acad Dermatol 1992; 27 (2 Pt 2): 348–355
Joh GY, Traupe H, Metze D, et al. A novel dinucleotide mutation in keratin 10 in the annular epidermolytic ichthyosis variant of bullous congenital ichthyosiform erythroderma. J Invest Dermatol 1997; 108 (3): 357–361
Suga Y, Duncan KO, Heald PW, et al. A novel helix termination mutation in keratin 10 in annular epidermolytic ichthyosis, a variant of bullous congenital ichthyosiform erythroderma. J Invest Dermatol 1998; III (6): 1220–1223
Sybert VP, Francis JS, Corden LD, et al. Cyclic ichthyosis with epidermolytic hyperkeratosis: a phenotype conferred by mutations in the 2B domain of keratin K1. Am J Hum Genet 1999; 64 (3): 732–738
Michael EJ, Schneiderman P, Grossman ME, et al. Epidermolytic hyperkeratosis with polycyclic psoriasiform plaques resulting from a mutation in the keratin 1 gene. Exp Dermatol 1999; 8 (6): 501–503
Siemens HW. Dichtung und Wahrheit über die die “Ichthyosis bullosa”, mit Bemerkungen zur Systematik der Epidermolysen. Arch Dermatol Syph (Berl) 1937; 175: 590–608
McLean WHI, Morley SM, Lane EB, et al. Ichthyosis bullosa of Siemens: a disease involving keratin 2e. J Invest Dermatol 1994; 103 (3): 277–281
Collin C, MoH R, Kubicka S, et al. Characterization of human cytokeratin 2, an epidermal cytoskeleton protein synthesized late during differentiation. Exp Cell Res 1992; 202: 132–141
Steijlen P, Kremer H, Vakilzadeh F, et al. Genetic linkage of the keratin type II gene cluster with ichthyosis bullosa of Siemens and with autosomal dominant ichthyosis exfoliativa. J Invest Dermatol 1994; 103: 282–285
Kremer H, Zeeuwen P, McLean WHI, et al. Ichthyosis bullosa of siemens is caused by mutations in the keratin 2e gene. J Invest Dermatol 1994; 103 (3): 286–289
Rothnagel JA, Traupe H, Wojcik S, et al. Mutations in the rod domain of keratin 2e in patients with ichthyosis bullosa of Siemens. Nat Genet 1994; 7 (4): 485–490
Gorlin RJ, Pindborg JJ, Cohen Jr MM. Syndromes of the Head and Neck. 2nd ed. New York: McGraw-Hill, 1976
Jadassohn J, Lewandowsky F. Pachyonychia congenita. Jacobs Ikonographia Dermatologica; vol 1. Berlin: Urban and Schwarzenberg, 1906
Jackson ADM, Lawler SD. Pachyonychia congenita: a report of six cases in one family. Ann Eugen 1951; 16: 142–146
McLean WHI, Rugg EL, Lunny DP, et al. Keratin 16 and keratin 17 mutations cause pachyonychia congenita. Nat Genet 1995; 9: 273–278
Smith FJD, Del Monaco M, Steijlen PM, et al. Novel proline substitution mutations in keratin 16 in two cases of pachyonychia conge nita type 1. Br J Dermatol 1999; 141: 1010–1016
Bowden PE, Haley JL, Kansky A, et al. Mutation of a type II keratin gene (K6a) in pachyonychia congenita. Nat Genet 1995; 10: 363–365
Lin MT, Levy ML, Bowden PE, et al. Identification of sporadic mutations in the helix initiation motif of keratin 6 in two pachyonychia congenita patients: further evidence for a mutational hot spot. Exp Dermatol 1999; 8 (2): 115–119
Terrinoni A, Smith FJD, Didona B, et al. Novel and recurrent mutations in the genes encoding keratins K6a, K16 and K17 in thirteen cases of pachyonychia congenita. J Invest Dermatol 2001; 117 (6): 1391–1396
Connors JB, Rahil AK, Smith FJD, et al. Delayed-onset pachyonychia congenita associated with a novel mutation in the central 2B domain of keratin 16. Br J Dermatol 2001; 144 (5): 1058–1062
Terrinoni A, Puddu P, Didona B, et al. A mutation in the V1 domain of K16 is responsible for unilateral palmoplantar verrucous nevus. J Invest Dermatol 2000; 114 (6): 1136–1140
Munro CS, Carter S, Bryce S, et al. A gene for pachyonychia congenita is closely linked to the keratin gene cluster on 17q12-q21. J Med Genet 1994; 31: 675–678
Troyanovsky SM, Guelstein VI, Tchipysheva TA, et al. Patterns of expression of K17 in human epithelia: dependency on cell position. J Cell Sci 1989; 93: 419–426
McGowan KM, Coulombe PA. Keratin 17 expression in the hard epithelial context of the hair and nail, and its relevance for the pachyonychia conge nita phenotype. J Invest Dermatol 2001; 114 (6): 1101–1107
Smith FJD, Coleman CM, Bayoumy NM, et al. Novel keratin 17 mutations in pachyonychia congenita type 2. J Invest Dermatol 2001; 116 (5): 806–808
Smith FJD, Jonkrnan MF, van Goor H, et al. A mutation in human keratin K6b produces a phenocopy of the K17 disorder pachyonychia congenita type 2. Hum Mol Genet 1998; 7 (7): 1143–1148
Covello SP, Smith Fill, Sillevis Smitt JH, et al. Keratin 17 mutations cause either steatocystoma multiplex or pachyonychia congenita type 2. Br J Dermatol 1998; 139: 475–480
Smith Fill, Corden ill, Rugg EL, et al. Missense mutations in keratin 17 cause either pachyonychia congenita type 2 or a phenotype resembling steatocystoma multiplex. J Invest Dermatol 1997; 108 (2): 220–223
Ratnavel RC, Griffiths W A. The inherited palmoplantar keratodermas. Br J Dermatol 1997; 137 (4): 485–490
Vorner H. Zur Kenntniss des Keratoma hereditarium palmare et plantare. Arch Derm Syph 1901; 56: 3–31
Navsaria HA, Swensson O, Ratnavel RC, et al. ULtrastructural-changes resulting from keratin-9 gene-mutations in 2 families with epidermolytic palmoplantar keratoderma. J Invest Dermatol 1995; 104 (3): 425–429
Reis A, Kuster W, Eckardt R, et al. Mapping of a gene for epidermolytic palmoplantar keratoderma to the region of acidic keratin gene cluster at 17q12-q21. Hum Genet 1992; 90: 113–116
Reis A, Hennies H-C, Langbein L, et al. Keratin 9 gene mutations in epidermolytic palmoplantar keratoderma (EPPK). Nat Genet 1994; 6: 174–179
Coleman CM, Munro CS, Smith Fill, et al. Epidermolytic palmoplantar keratoderma due to a novel type of keratin mutation, a 3 bp insertion in the keratin 9 helix termination motif. Br J Dermatol 1999; 140: 486–490
Hatsell SJ, Eady RA, Wennerstrand L, et al. Novel splice site mutation in keratin 1 underlies mild epidermolytic palmoplantar keratoderma in three kindreds. J Invest Dermatol 2001; 116 (4): 606–609
Kimonis V, DiGiovanna JJ, Yang J-M, et al. A mutation in the VI end domain of keratin 1 in non-epidermolytic palmar-plantar keratoderma. J Invest Dermatol 1994; 103 (6): 764–769
Sprecher E, Ishida-Yamamoto A, Becker OM, et al. Evidence for novel functions of the keratin tail emerging from a mutation causing ichthyosis hystrix. J Invest Derrnatol 2001; 116 (4): 511–519
Bonifas JM, Bare JW, Chen MA, et al. Evidence against keratin gene mutations in a family with ichthyosis hystrix Curth-Macklin. J Invest Dermatol 1993; 101 (6): 890–891
Shamsher MK, Navsaria HA, Stevens HP, et al. Novel mutations in keratin 16 gene underly focal non-epidermolytic palmoplantar keratoderma (NEPPK) in 2 families. Hum Mol Genet 1995; 4 (10): 1875–1881
Smith FJD, Fisher MP, Healy E, et al. Novel keratin 16 mutations and protein expression studies in pachyonychia congenita type 1 and focal palmoplantar keratoderma. Exp Dermatol 2000; 9: 170–177
Jorgenson RJ, Levin S. White sponge nevus. Arch Dermatol 1981; 117: 73–76
McGinnis JP, Turner JE. Ultrastructure of the white sponge nevus. Oral Surg 1975; 40 (5): 644–651
Frithiof L, Banoczy J. White sponge nevus (leukoedema exfoliativum mucosae oris): ultrastructural observations. Oral Surg 1976; 41 (5): 607–622
Rugg EL, McLean WHI, Allison WE, et al. A mutation in the mucosal keratin K4 is associated with oral white sponge nevus. Nat Genet 1995; 11: 450–452
Richard G, DeLaurenzi V, DiDona B, et al. Keratin-13 point mutation underlies the hereditary mucosal epithelial disorder white sponge nevus. Nat Genet 1995; 11 (4): 453–455
Terrinoni A, Candi E, Oddi S, et al. A glutamine insertion in the 1 A alpha helical domain of the keratin 4 gene in a familial case of white sponge nevus. J Invest Derrnatol 2000; 114 (2): 388–391
Terrinoni A, Rugg EL, Lane EB, et al. A novel mutation in the keratin 13 gene causing oral white sponge nevus. J Dent Res 2001; 80 (3): 919–923
Rugg E, Magee G, Wilson N, et al. Identification of two novel mutations in keratin 13 as the cause of white sponge naevus. Oral Dis 1999; 5 (4): 321–324
Munro CS. The clinical consequences of keratin gene defects. Dermatol Practice 1998; 6 (6): 8–11
Meesmann A, Wilke F. Klinische und anatomische Untersuchungen ueber eine bisher unbekannte, dominant vererbte Epitheldystrophie der Hornhaut. Klin Mbl Augenheilk 1939; 103: 361–391
Tremblay M, Dube I. Meesmann’s corneal dystrophy: ultrastructural features. Can J Ophthalmol 1982; 17 (1): 24–28
Liu CY, Zhu G, Westerhausen-Larson A, et al. Cornea-specific expression of K12 keratin during mouse development. Curr Eye Res 1993; 12 (11): 963–974
Kao WW-Y, Liu C-Y, Converse RL, et al. Keratin 12 deficient mice have fragile corneal epithelia. Invest Ophthalmol Vis Sci 1996; 37: 2572–2584
Irvine AD, Corden LD, Swensson O, et al. Mutations in cornea-specific keratins K3 or K12 cause Meesmann’s corneal dystrophy. Nat Genet 1997; 16: 184–187
Nishida K, Honma Y, Dota A, et al. Isolation and chromosomal localization of a cornea-specific human keratin 12 gene and detection of four mutations in Meesmann corneal epithelial dystrophy. Am J Hum Genet 1997; 61 (6): 1268–1275
Coleman CM, Hannush S, Covello SP, et al. A novel mutation in the helix termination motif of keratin K12 in a US family with Meesmann corneal dystrophy. Am J Ophthalmol 1999; 128 (6): 687–691
Corden LD, Swensson O, Swensson B, et al. Molecular genetics of Meesmann’s corneal dystrophy: ancestral and novel mutations in keratin 12 (K12) and complete sequence of the human KRT12 gene. Exp Eye Res 2000; 70 (1): 41–49
Corden LD, Swensson O, Swensson B, et al. A novel keratin 12 mutation in a German kindred with Meesmann’s corneal dystrophy. Br J Ophthalmol 2000; 84 (5): 527–530
Ku NO, Michie S, Oshima RG, et al. Chronic hepatitis, hepatocyte fragility, and increased soluble phosphoglycokeratins in transgenic mice expressing a keratin 18 conserved arginine mutant. J Cell Biol 1995; 131 (5): 1303–1314
Ku NO, Wright TL, Terrault NA, et al. Mutation of human keratin 18 in association with cryptogenic cirrhosis. J Din Invest 1997; 99 (1): 19–23
Ku NO, Gish R, Wright TL, et al. Keratin 8 mutations in patients with cryptogenic liver disease. N Engl J Med 2001; 344 (21): 1580–1587
Ito M, Hashimoto K, Katsuumi K, et al. Pathogenesis of monilethrix: computer stereography and electron microscopy. J Invest Dermatol 1990; 95 (2): 186–194
Healy E, Holmes SC, Belgaid CE, et al. A gene for monilethrix is closely linked to the type II keratin gene cluster at 12gB. Hum Mol Genet 1995; 4 (12): 2399–2402
Stevens HP, Kelsell DP, Bryant SP, et al. Linkage of monilethrix to the trichocyte and epithelial keratin gene cluster on 12q11-q13. J Invest Dermatol 1996; 106 (4): 795–797
Winter H, Rogers MA, Langbein L, et al. Mutations in the hair cortex keratin hHb6 cause the inherited hair disease monilethrix. Nat Genet 1997; 16 (4): 372–374
Winter H, Rogers MA, Gebhardt M, et al. A new mutation in the type II hair cortex keratin hHbl involved in the inherited hair disorder monilethrix. Hum Genet 1997; 101 (2): 165–169
Takahashi K, Paladini RD, Coulombe PA. Cloning and characterization of multiple human genes and cDN As encoding highly related type II keratin 6 isoforms. J Biol Chem 1995; 270 (31): 18581–18592
Savtchenko ES, Freedberg IM, Choi I-Y, et al. Inactivation of human keratin genes: the spectrum of mutations in the sequence of an acidic keratin pseudogene. Mol Biol Evol 1988; 5 (1): 97–108
Troyanovsky SM, Leube RE, Franke WW. Characterization of the human gene encoding cytokeratin 17 and its expression pattern. Eur J Cell Biol 1992; 59 (1): 127–137
Ruud P, Fodstad O, Hovig E. Identification of a novel cytokeratin 19 pseudogene that may interfere with reverse transcriptase-polymerase chain reaction assays used to detect micro metastatic tumor cells. Int J Cancer 1999; 80 (1): 119–125
RayChaudhury A, Marchuk D, Lindhurst M, et al. Three tightly linked genes encoding human type I keratins: conservation of sequence in the 5’-untranslated leader and 5’-upstream regions of coexpressed keratin genes. Mol Cell Biol 1986; 6: 539–548
Paladini RD, Takahashi K, Gant TM, et al. cDNA cloning and bacterial expression of the human type I keratin 16. Biochem Biophys Res Commun 1995; 215 (2): 517–523
Smith FJD, McKusick V A, Nielsen K, et al. Cloning of multiple keratin 16 genes facilitates prenatal diagnosis of pachyonychia congenita type 1. Prenat Diagn 1999; 19: 941–946
Smith FJD, McKenna KE, Irvine AD, et al. A mutation detection strategy for the human K6A gene and novel mutations in two cases of pachyonychia congenita type 1. Exp Dermatol 1999; 8: 109–114
McLean Will, Pulkkinen L, Smith FJD, et al. Loss of plectin causes epidermolysis bullosa with muscular dystrophy: cDNA cloning and genomic organisation. Genes Dev 1996; 10: 1724–1735
Uitto I, Pulkkinen L, Smith FJD, et al. Plectin and human genetic disorders of skin and muscle: the paradigm of epidermolysis bullosa with muscular dystrophy. Exp Dermatol 1996; 5: 237–246
McGrath JA, McMillan JR, Shemanko CS, et al. Mutations in the plakophilin 1 gene can result in ectodermal dysplasia/skin fragility syndrome. Nat Genet 1997; 17 (2): 240–244
Cao T, Longley MA, Wang XJ, et al. An inducible mouse model for epidermolysis bullosa simplex: implications for gene therapy. J Cell Biol 2001; 152 (3): 651–656
Rothnagel JA, Longley MA, Holder RA, et al. Prenatal diagnosis of epidermolytic hyperkeratosis by direct gene sequencing. J Invest Dermatol 1994; 102 (1): 13–16
Rothnagel JA, Lin MT, Longley MA, et al. Prenatal diagnosis for keratin mutations to exclude transmission of epidermolytic hyperkeratosis. Prenat Diagn 1998; 18 (8): 826–830
Acknowledgements
Thanks to the many patients and their clinicians who have helped our research efforts over the years. Thanks to Pamela Wood, Dr Alan Irvine and Dr Colin Munro for providing illustrations, to Dr Irwin McLean for critical reading of the manuscript and to Andrew Cassidy for Intermediate Filament Database consultation. The author is currently supported by the Wellcome Trust (Senior Research Fellowship grant held by WHI McLean). Research in the Epithelial Genetics Group is also supported by DEBRA, UK.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Smith, F.J.D. The Molecular Genetics of Keratin Disorders. Am J Clin Dermatol 4, 347–364 (2003). https://doi.org/10.2165/00128071-200304050-00005
Published:
Issue Date:
DOI: https://doi.org/10.2165/00128071-200304050-00005