
Abstract
Skeletal ciliopathies are a heterogeneous group of autosomal recessive osteochondrodysplasias caused by defects in formation, maintenance and function of the primary cilium. Mutations in the underlying genes affect the molecular motors, intraflagellar transport complexes (IFT), or the basal body. The more severe phenotypes are caused by defects of genes of the dynein-2 complex, where mutations in DYNC2H1, WDR34 and WDR60 have been identified. In a patient with a Jeune-like phenotype we performed exome sequencing and identified compound heterozygous missense and nonsense mutations in DYNC2LI1 segregating with the phenotype. DYNC2LI1 is ubiquitously expressed and interacts with DYNC2H1 to form the dynein-2 complex important for retrograde IFT. Using DYNC2LI1 siRNA knockdown in fibroblasts we identified a significantly reduced cilia length proposed to affect cilia function. In addition, depletion of DYNC2LI1 induced altered cilia morphology with broadened ciliary tips and accumulation of IFT-B complex proteins in accordance with retrograde IFT defects. Our results expand the clinical spectrum of ciliopathies caused by defects of the dynein-2 complex.
Similar content being viewed by others


Introduction
The primary cilium is a nearly ubiquitous organelle of non-proliferative vertebrate cells. This organelle functions as an antenna to sense extracellular stimuli via various ciliary membrane receptors and transmits these signals into the cell to initiate intracellular transduction cascades of different signaling pathways1,2,3,4. These include the Hedgehog, Wnt, planar cell polarity, FGF, Notch, mTor, PDGF and the Hippo signaling pathways5,6,7,8,9,10,11,12,13.
Cilia play important roles in differentiation, migration, proliferation, determination of left-right asymmetry and are thus important for the embryonic and postnatal development and proper organ function in adulthood1,14,15. The non-motile primary cilium consists of two main components, the basal body complex on the cytoplasmic side of the cell membrane and the ciliary axoneme, surrounded by a cytoplasmic membrane extending into the extracellular space. The axoneme is radially arranged by nine microtubule doublets growing out of the distal end of the basal body (9 × 2 + 0 structure). The basal body complex consists of a mother and daughter centriole embedded in the pericentriolar material (PCM)3,4,14,16,17. The junction between the basal body and the axoneme, the transition zone, is a specialized structure with a barrier function (“ciliary gate”), regulating the passage of ciliary proteins into or out of the primary cilium18,19.
During the cell cycle cells present a primary cilium in G0/G1 phase and before entering mitosis the cilium is disassembled. For assembly, maintenance and dismantling different proteins are required, brought to their destination via the intraflagellar transport (IFT). The IFT is a bidirectional transit system that transports cargos to the tip of the cilium (anterograde transport) and back to the base (retrograde transport)17,20,21. Kinesin-2 motors and subunits of the IFT-B complex are integral parts of the anterograde movement, whereas cytoplasmic dynein-2 and IFT-A components are responsible for retrograde transport16,22,23. Cell cycle regulatory signals, cytoplasmic vesicle transport systems and recruitment of all required IFT components at the right time and the right place are important for proper ciliogenesis.
Defects of genes encoding a variety of proteins involved in cilia formation, maintenance and function, such as IFT subcomponents and components of the ciliary axoneme, transition zone or basal body, lead to the broad phenotypic spectrum of ciliopathies15,24. Many of the associated phenotypes include brain malformations, polydactyly, kidney cysts, retinal degeneration and skeletal abnormalities. Today, mutations in 14 different genes have been identified to be causative for skeletal ciliopathies. Because of the genetic heterogeneity of skeletal dysplasias, disrupted proteins affect either the dynein motor (DYNC2H1), or the core components of the IFT transport complexes (IFT43, IFT80, IFT122, IFT140, IFT172, WDR19, WDR34, WDR35, WDR60 and TTC21B) or the basal body region (NEK1, EVC, EVC2)25,26,27,28,29,30. The phenotypic spectrum of skeletal ciliopathies includes short ribs, narrow thorax, short stature, postaxial polydactyly and other skeletal abnormalities. These phenotypes are shared by the skeletal short-rib thoracic dysplasias (SRTD 1 - 12 [MIM 208500, 611263, 613091, 613819, 614376, 263520, 614091, 615503, 266920, 615630, 615633, 269860]), Ellis-van Crefeld syndrome (EVC [MIM 225500]), cranioectodermal dysplasias (CED1-4 [MIM 218330, 613610, 614099, 614378]) and Weyers acrofacial dysostosis (WAD [MIM 193530])25,31,32,33.
Here, we performed exome sequencing in a family with a Jeune-like intermediate phenotype (SRTD and EVC overlapping clinical findings) and identified compound heterozygous nonsense and missense mutations in the DYNC2LI1 gene. DYNC2LI1 is known as a component of the dynein-2 complex important for retrograde IFT34,35. Knockdown experiments resulted in shorter cilia with abnormally bulged tips, similar to other ciliopathies with retrograde IFT defects. Mutations in the light intermediate chain of the dynein-2 complex are further expanding the clinical spectrum of skeletal ciliopathies.
Results
Clinical features of patients with DYNC2LI1 mutations
The affected children were the offsprings of non-consanguineous parents (Supplementary Fig. 1a). The first child (patient 1) developed a respiratory distress syndrome and died at the age of 3 days. Three further pregnancies ended as missed abortions. The second child (patient 2) developed well, but showed dysmorphic features. During the following pregnancy fetal postaxial polydactyly was noticed and the pregnancy terminated at 19th week of gestation (patient 3). The observed clinical features were classified between Ellis-van-Crefeld syndrome and Jeune syndrome. Although the affected children present with variable features they do share characteristics in common.
Patient 1 showed a polyhydramnion during pregnancy and was born at 38th weeks of gestation with a weight of 3300 g (0.3 s.d.) and a body length of 50 cm (–0.17 s.d.). Clinical findings included agenesis of the epiglottis, postaxial hexadactyly of the both hands and the right foot, hypoplastic nails, atrioventricular septal defect and hepatosplenomegaly (Supplementary Fig. 1b-d). She presented with distinctive facial features characterized by medial cleft lip, low set ears, asymmetric tongue, hyperplastic gingivae, epicanthal fold and down-slanting palpebral fissures. A hypoplastic thorax led to a respiratory distress syndrome and the patient died two days after birth. Her karyotype was normal (46,XX).
Patient 2 also showed a polyhydramnion during pregnancy and was born at 40th week of gestation with a weight of 4120 g (1.53 s.d.) and a body length of 53 cm (0.59 s.d.). Her psychomotor development was normal. She spoke first words at 8 month and was able to walk independently at 14 month. She likewise presented with postaxial hexadactyly of the left hand, brachydactyly, a small medial cleft lip, a hypoplastic epiglottis and narrow thorax, in addition we observed a syndactyly VI/VII of the right hand (Supplementary Fig. 1e-g). Her facial feature include a broad and prominent forehead, a depressed nasal bridge, broad and up-slanting nasal tip and low set ears. She also went through respiratory distress syndrome but recovered after 2 month. Beside an episode of pneumonia she showed no signs of increases susceptibility to infections. No further malformation are known.
Patient 3 was the fetus after termination of the pregnancy at 19th week of gestation. During pregnancy polyhydramnion and postaxial hexadacytly was noticed (Supplementary Fig. 1h-j). In addition, the fetus presented with a medial cleft lip and a pronounced narrow thorax.
Radiological investigation of both girls were performed and revealed cone-shaped epiphysis of the phalangeal bones, a metaphyseal dysplasia and a narrow thorax with short ribs (personal communication with Prof. F. Majewksi), so an atypical Jeune syndrome was proposed (Supplementary Fig. 1k,l).
Whole exome sequencing and candidate screening
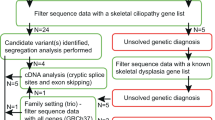
Exome sequencing has been demonstrated to be highly successful to identify the underlying genetic cause of autosomal recessive inherited entities36,37,38,39,40,41. After Agilent sure select capturing and exome sequencing of patient 2 we received 156.9 M single end reads. 85.01% of the reads were mapped to the human genome version hg19 and 88.06% were on-target. 81.5% of the captured target was covered at least 5x with an average depth of 73.3-fold. A total of 52,679 variants were called, of which 2,076 were indels and 50,603 were single nucleotide variants. Based on the supposed rare incidence of the phenotype we excluded all frequent variants (above 0.01%), detected in any of the 666 in-house control individuals as well as all annotated variants of dbSNP132, the 1000genomes project (phase1) and the exome variant server (http://evs.gs.washington.edu/EVS). Excluding intergenic, intronic and synonymous variants and proposing an autosomal recessive inheritance led to 58 remaining, variants of unknown significance (VUS) in 4 genes. Based on the mutational effect of these variants, their conservation (PhyloP, GERP++) and prediction program scores of SIFT, PolyPhen2 and MutationTaster, CADD only the two DYNC2LI1 mutations remained as candidate variants (Supplementary Table 1): A nonsense mutation c.622C > T (p.Arg208Ter; NM_001193464) in exon 8 and the missense mutation c.662C > T (p.Thr221Ile; NM_001193464) in exon 9 of the DYNC2LI1 gene. Nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence, with the initiation codon as codon 1. Both variants could be confirmed by Sanger sequencing (Supplementary Fig. 2) and were not present in 858 control chromosomes of healthy individuals and in the 61,486 exomes of the Exome Aggregation Consortium (Exome Aggregation Consortium (ExAC), Cambridge, MA (URL: http://exac.broadinstitute.org) Nov. 2014). The affected positions are highly conserved on nucleotide and amino acid level. No mutations in any of the known skeletal ciliopathy genes or other potential candidate genes were identified in this individual. The healthy mother, brother and son were carrier for the missense mutation p.Thr221Ile. DNA of the healthy father and the affected patients 2 and 3 was not available. Based on the family history the nonsense mutation is paternally inherited. Therefore, the identified mutations are highly likely to be pathogenic.
DYNC2LI1 expression
We were not able to investigate the effect of the mutations on the expression level, as RNA of the patients was not available. We analyzed the expression levels of DYNC2LI1 in different tissues with adult and fetal complementary DNA (cDNA) panels as well as in osteoblasts and chondrocytes. We examined a ubiquitous expression of DYNC2LI1 in all examined tissues (Supplementary Fig. 3). However, the overall highest expression levels were measured in chondrocytes, brain and kidney, no differences between fetal and adult tissues were observed except for a higher fetal expression in brain, kidney and lungs. This is in accordance with expression profiles of genes involved in structure and function of the primary cilium. In addition, we observed an expression of all 3 isoforms in the examined tissues with the highest contribution of the 2 longest isoforms.
Domain structure and structural effects of DYNC2LI1 mutations
To date, structural information of DYNC2LI1 remains elusive, but comparison of the protein sequence (UniProt Q8TCX1-1) with known motifs predicted a P-loop containing nucleoside triphosphate hydrolases domain (position aa 30 – 240) including a Dynein light intermediate chain domain (position aa 30 – 163; http://www.ebi.ac.uk/interpro) (Fig. 1a). The most common functions of the P-loop containing nucleoside triphosphate hydrolases domains are the hydrolysis of nucleoside triphosphate42. Modelling of the nucleoside triphosphate hydrolase domain revealed that Thr221 (Fig. 1a,b) is close to the nucleoside binding site. An exchange from threonine to isoleucine in the mutant causes steric clashes and decreases the site of the nucleoside binding pocket (Fig. 1b). Therefore, the mutation is expected to impair the hydrolase function of DYNC2LI1.

Identified mutations in DYNC2LI1 and protein model.
(a) Compound heterozygous missense and nonsense mutation are located in the nucleoside triphosphate hydrolase domain of DYNC2LI1. (b) Model of the nucleoside triphosphate hydrolase domain of DYNC2LI1 in the wild-type and p.Thr221Ile mutant. The protein backbone is depicted as cyan tube and residue 221 is shown in space-filled presentation and colored according to the atom types. A bound nucleoside diphosphate (GDP) is shown as sticks. Alternatively, ADP may be bound at this site, which cannot be safely discriminated at the present resolution of the model. (c) Scheme of the primary cilium and known mutations in skeletal ciliopathies associated genes (EVC/EVC2 = Ellis-van Crefeld syndrome and WAD; NEK1 = SRTD6; TTC21B = SRTD4; IFT80 = SRTD2; IFT43 = CED3; IFT122 = CED1; IFT140 = SRTD9; IFT172 = SRTD10; WDR19 = SRTD5 and CED4; WDR35 = SRTD7 and CED2; WDR34 = SRTD11; WDR60 = SRTD8; DYNC2H1 = SRTD3; DYNC2LI1 = novel intermediate phenotype of our patients)25,26,27,28,29,30,34. Abbreviations: WAD (Weyers acrofacial dysostosis), CED (cranioectodermal dysplasia), SRTD (short-rib thoracic dysplasia).
As RNA was not available, we were not able to test whether the nonsense mutation might lead to mRNA decay or a shorter protein. However, a predicted truncated protein will miss the coiled-coil domain (position aa 315 – 340) which might be necessary for interaction with the coiled coil domain of DYNC2H1 of the dynein-2 complex.
DYNC2LI1 localizes to the primary cilium and DYNC2LI1 defects affect morphology and cilia length
As the dynein-2 complex is part of the retrograde IFT of the primary cilium we expected the DYNC2LI1 protein to be localized to the primary cilium (Fig. 1c)35. Using immunofluorescence analysis in ciliated fibroblast cells we confirmed the localization of the DYNC2LI1 protein to the cytoplasm, centrosomes, as well as around the basal body and the transition zone of the primary cilium (Fig. 2). To determine the patient’s phenotype on a cellular level we performed immunofluorescence staining on human fibroblasts. As patient-derived fibroblasts were not available, the cellular effects of DYNC2LI1 depletion were examined following siRNA knockdown of DYNC2LI1 and compared to scrambled siRNA. The remaining expression rate of DYNC2LI1 was on average 39%. Cilia were examined after siRNA transfection and following 5 days of serum starvation to induce ciliogenesis. While cilia were detected in the majority of the cells, no significant difference in percentage of ciliated cells was detected (Fig. 3 a,b: 85% ciliated cells in scrambled controls vs. 84% in DYNC2LI1 depleted cells, p = 0.927 χ2-test). As DYNC2LI1 is part of the dynein-2 complex and defects of other components of this complex have been associated with alterations of cilia length and morphology we performed further immunofluorescence analysis of the DYNC2LI1 depleted cells. Control cilia showed a mean cilia length of median 1.83 μm, in comparison to cilia in DYNC2LI1 depleted cells, which were only 1.42 μm long (Fig. 3 c,d). Measured differences were highly significant with a p-value of 1.205 × 10−6 [Mann-Whitney-U test]. In addition, we observed significantly altered cilia morphology with broadened ciliary tips in 14% of cilia of the DYNC2LI1 depleted cells, compared to 6% abnormal ciliary structures in scrambled controls [p = 0.028 χ2-test ] (Fig. 4 a). To study the defect in retrograde IFT due to the abnormal ciliary morphology, we stained components of the IFT-B complex IFT57 and IFT88. We observed a localization of these at the basal body region of the primary cilium in scrambled controls and knockdown cells. Morphological abnormal cilia in DYNC2LI1 depleted cells showed an accumulation of IFT57 and IFT88 in the bulbous ciliary tip confirming a retrograde IFT defect in these cells (Fig. 4 b,c).

Localization of the DYNC2LI1 protein in control fibroblasts.
Immunofluorescence staining of 5 days starved control fibroblasts with antibodies against polyglutamylated tubulin (ciliary axoneme, red), DYNC2LI1 (green) and DAPI (DNA, blue in merge) identified localization of DYNC2LI1 to the basal body region (a) and to the transition zone (b) of the primary cilium, as well as to the centrosomes (c) during mitosis. Magnifications are shown in white boxes, white scale bars 10 μm.

Number of primary cilia and cilia length in DYNC2LI1 depleted cells compared to scrambled control.
Immunofluorescence staining of 5 days starved fibroblasts (scrambled control vs. DYNC2LI1siRNA) with antibodies against polyglutamylated tubulin (primary cilium, red), PCNT (green) and DAPI (DNA, blue in merge). (a,b) No significant differences in the number of cilium presenting cells were observed, 85% of control cells and 84% of DYNC2LI1 depleted cells showed primary cilium formation (p = 0.927 [χ2-test], white scale bars 20 μm). (c,d) The median cilia length was 1.83 μm in scrambled control cells, whereas cilia in knockdown cells were significantly shorter with median of 1.42 μm (p = 1.205 × 10−6 [Mann-Whitney-U test]). Magnifications are shown in white boxes, white scale bars 10 μm).

DYNC2LI1 defect cells present with abnormal cilia morphology confirmed retrograde IFT defects.
Immunofluorescence staining of 5 days starved fibroblasts (scrambled control vs. DYNC2LI1siRNA) with antibodies against polyglutamylated tubulin (primary cilium, red), PCNT/IFT57/IFT88 (green) and DAPI (DNA, blue in merge). (a) DYNC2LI1 depleted cells showed in addition to the reduced ciliary length further ciliary morphological abnormalities with bulbous tips in 14% of analyzed cilia. Scrambled control cells present normal cilia structure, but we observed as well malformations in 6% of measured control cilia (p = 0.028 [χ2-test]). (b) Scrambled controls and knockdown cells showed a localization of IFT57 at the basal body region of the primary cilium. In addition, knockdown of DYNC2LI1 resulted in an accumulation of IFT57 - component of IFT-B complex - in the bulbous ciliary tip (arrows). (c) IFT88 - another component of the IFT-B complex - is localized at the basal body region of the primary cilium in scrambled controls and knockdown cells. IFT88 is present at the ciliary tip in scrambled controls compared to a strong accumulation in the bulbous tip of DYNC2LI1 depleted cells (arrows). White scale bars 2.5 μm, three-dimensional reconstruction was performed using Imaris software (Bitplane, Zurich).
Discussion
The primary cilium has a pivotal role in a variety of complex processes during pre- and postnatal development of almost all vertebrate cells1,14,15. The assembly, maintenance and disassembly of the primary cilium is highly controlled throughout the cell cycle to ensure proper function of the associated pathways43. Several disorders have been associated with defects of the primary cilium15,24,31. The observed phenotypes of these ciliopathies represent alteration of the ciliary pathways and can affect nearly every major organ44. Thus, these clinical entities have many clinical features in common. This holds true in particular for the group of the skeletal ciliopathies. Here, the most common group constitutes of the short-rib thoracic dysplasias (SRTDs), including the asphyxiating thorax dysplasia / Jeune and Ellis-van Crefeld syndrome25,32. Jeune syndrome / ATD is distinguished by severely constricted thorax leading to respiratory insufficiency, shortened tubular bones and inconsistent polydactyly45,46,47. Features observed in Elli-van Crefeld syndrome are congenital cardiac defects, dysplastic nails and teeth and labio-gingival adhesions, as well as skeletal features like polydactyly, short ribs and limbs48. These entities show significant clinical overlap and variable phenotypical expression. The affected individuals of the family reported here present with clinical features that include hypoplastic thorax, short stature, polydactyly, hypoplastic nails and atrioventricular septal defects. In accordance with the taxonomy, our patients have been classified as Jeune-like or EVC.
NGS has been proven to be effective to identify the underlying genetic cause of inherited diseases36,37,38,39,40,41. Concerning the genetic heterogeneity of the skeletal ciliopathies we aimed at the identification of the underlying cause in our family by using whole exome sequencing after SureSelect enrichment. After excluding mutations in the known genes associated with skeletal ciliopathies we focused on rare, coding variants in cilia associated genes5,49,50,51,52. This identified the two heterozygous mutations c.622C > T (p.Arg208Ter) in exon 8 and c.662C > T (p.Thr221Ile) in exon 9 of the DYNC2LI1 gene (Fig. 1 a). In agreement with autosomal-recessive inheritance the non-affected family members were heterozygous carriers for the identified mutations. These mutations were confirmed by Sanger sequencing and could be excluded in further control samples. Both mutations were not present in the 61,486 exomes of the Exome Aggregation Consortium and are therefore conserved, rare variants. Mice homozygous for a knockout of the orthologous gene Dync2li1tm1Aar are not viable and die before embryonic day 11.553. These mice lack monocilia and develop neural tube and cardiac defects and defects in trunck and tail development. Thus, we considered these mutations as most likely disease causing.
DYNC2LI1 (D2LIC, LIC3) consists of 3 isoforms (NM_001193464; NM_016008; NM_015522) with the longest to be composed of 13 exons encoding a 352-amino-acid-long protein. It is ubiquitously expressed with high expression levels in brain, kidney, lung and testes shown in immunoblots of adult mice54. In human tissues we confirmed the highest mRNA expression levels in chondrocytes, brain and kidney (Supplementary Fig. 3). Significant difference with higher expression levels in fetal tissues was observed in brain, lung and kidney. The two longest isoforms are predominantly expressed in all examined tissues. As our mutations are located in these 2 longest isoforms only, we considered these to have a pathogenic effect on skeletal development in our patients and therefore to be disease causing.
DYNC2LI1 has been identified as part of the dynein-2 complex involved in the retrograde transport (retrograde IFT)34,35. Loss of cytoplasmic dynein-2 in dync2-li1 zebrafish morphants leads to shorter and lower amount of cilia with accumulation of IFT88 near the distal end of the connecting cilium55. These morphants have a characteristic phenotype with shortening of the body length, small eyes and kidney cysts in 62% as observed in ciliopathies. Interestingly, defects of other components of the dynein-2 complex, DYNC2H1, WDR34, WDR60 in human have been associated with the clinical phenotype of SRPS type III, Jeune syndrome / asphyxiating thoracic dystrophy (SRTD3/8/11)34.
Previous studies localized DYNC2LI1 in mammalian cells at the Golgi apparatus and the centrosomal region54,56,57. In addition, a localization to the axoneme of ciliated epithelia cells was reported. Our immunocytochemistry using an antibody against DYNC2LI1 supported the localization to the centrosome (Fig. 2c). Moreover, we refined the reported localization in the axoneme to be concentrated at its base and at the region of the transition zone (Fig. 2a,b). Even though Madhivanan et al.58 speculated that defects of proteins involved in the control of vesicle trafficking to the cilium leads to a more severe phenotype than defects of the IFT only, the pronounced clinical phenotypes of patients with dynein-2 complex defects, Jeune and our patient, do not support this observation.
On the basis of the observed ciliary defects caused by dysfunction of IFT and basal body proteins, we expected loss of function of DYNC2LI1 to cause morphological abnormal and missing cilia in humans. As we were not able to obtain patient cells, we used immunofluorescence in DYNC2LI1 depleted human fibroblasts. We did not observe a significant difference in the percentage of ciliated cells (Fig. 3a,b), a significant average size reduction could be detected (Fig. 3c,d). The observation of shorter cilia confirmed the results in zebrafish morphants and Dync2li1tm1Aar knockout mice53,55. Reduced cilia length with bulbous tip was although reported for defects of DYNC2H1 and WDR34, other components of the dynein-2 complex28,59. However, one report showed no significant difference in proportion of ciliated cells and cilia length in Wdr34-knockdown ATDC5 lines29. In a further component of the dynein-2 complex, WDR60, a reduced amount of ciliated cells but normal cilia length was observed in cell lines of affected patients30. Together, these data indicate that the reduction of ciliated cells as well as of cilia length is a common hallmark of dynein-2 defects. In addition, we showed a higher incidence of cilia with a bulbous tip suggesting a defect in microtubule architecture (Fig. 4 a). This was also observed in Dync2li1tm1Aar knockout mice where stumpy nodal cilia contain disorganized microtubules, IFT proteins and cellular debris53,60. IFT-B proteins to be accumulated in cilia with retrograde IFT transport defects include IFT57 and IFT8830,61. Morphological abnormal cilia in our knockdown cells showed a significant accumulation at the ciliary tip confirming the IFT transport defect in DYNC2LI1 deficient cells (Fig. 4 b,c).
As primary cilia act as sensors for different pathways, the defect of the dynein-2 complex leading to retrograde IFT impairment is consistent with impairment of these signaling cascades. Among others, defects of the Sonic Hedgehog pathway have been associated with limb-development defects confirming the skeletal phenotype in our patients24,62,63.
In conclusion, mutations in members of the dynein-2 complex result in Jeune syndrome / ATD and other forms of the SRTD spectrum. Different defects of this complex suggest a correlation between the amount of ciliated cells, the resulting cilia length and the functional IFT components with the phenotypically overlapping, but variable clinical features. Thus, the identification of mutations in DYNC2LI1 in our patients expands the phenotypic spectrum of skeletal ciliopathies.
Methods
Subjects and clinical data
Informed consent was obtained from the patients for experimental protocols and displaying participants’ facial appearances in publications. Peripheral blood samples were obtained from the patients as well as control individuals. Genomic DNA from all collected samples was extracted according to standard procedures. This study was approved by the Ethical Review Board of the Friedrich-Alexander-Universität Erlangen-Nürnberg.
Whole Exome Sequencing
DNA of patient 2 was enriched using the Agilent’s SureSelect Human All Exon Kit V3 (Agilent technologies, Santa Clara, CA) and sequenced in single-end reads on a SOLiD system (Life Sciences, Santa Clara, CA). Sequenced reads were mapped with LifeScope (Life Technologies, Carlsbad, CA) to the reference human genome assembly hg19 (GRCh37). Genotypes were additionally called with GATK 2 and SAMtools64,65,66. For variant annotation ANNOVAR67 (http://www.openbioinformatics.org/annovar) was used to check on gene information, substitutions of amino acids, predictions of SIFT v. 1.03 (http://sift.jcvi.org/)68, PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/)69 and MutationTaster (http://www.mutationtaster.org/)70, entries of dbSNP132, as well as allele frequencies of the 1000 Genomes Project (http://www.1000genomes.org)71 and NHLBI-ESP 5400. An in-house database consisting of 666 sequenced exomes was also used to exclude sequence errors and frequent variants. Graphical presentation of the mapped sequences was viewed with the Integrative Genomics Viewer (IGV)72,73. Variants were selected based on autosomal recessive inheritance, their population frequency, the location within the coding sequence (exon, splice site) and the functional effect. The resulting variants were Sanger sequenced and the segregation in the family tested
Protein modelling
The domain architecture of DYNC2LI1 was investigated using the InterPro74 and Genesilico75 Meta prediction servers. Molecular modelling of residues 28-241 of DYNC2LI1 was performed with Modeller 9.976 using the crystal structure of human RAB5B in complex with GDP (PDB code: 2HEI) as a template. The mutation was modeled using SwissModel77 and RasMol was used for structure analysis and visualization78.
DYNC2LI1 expression analysis in human tissues
Relative DYNC2LI1 expression was measured by quantitative real-time PCR using predesigned TaqMan Gene Expression Assays with the TaqMan Gene Expression Mastermix (Life Technologies) specific for all 3 isoforms (Hs00602913_m1, spanning exon 5-6), isoform 4 (Hs01005273_m1, spanning exon 6-7 of NM_001193464) and isoform 1 (Hs01011588_m1, spanning exon of 6-7 NM_016008). An isoform 2 (NM_015522) specific probe was designed using the Custom TaqMan Assay Design Tool (Life Technologies) for the target region chr2:44021785-44022314 (3’ UTR).
Expression levels were quantified in quadruplicates for each gene using the ΔΔCt method with four endogenous control genes (Beta-Actin [huACTB], Beta-2-Microglobulin [huB2M], acidic ribosomal protein [huPO], phosphoglycerate kinase 1 [huPGK1]).
We used commercially available cDNA panels (Human Fetal MTCTM Panel”, “Human MTCTM Panel I” [Clonetech, Mountain View, USA]) covering fetal (whole brain, heart, liver, lung, kidney) and adult (whole brain, heart, liver, lung, kidney) tissues to determine the expression pattern of DYNC2LI1 in different human tissues. RNA of osteoblasts and chondrocytes were obtained from 2 different (osteoblasts) and 8 different (chondrocytes) adult individuals.
Cell culture
Primary fibroblasts of a healthy control were cultured using Dulbecco’s modified Eagle medium (DMEM, Life Technologies), patients fibroblasts were not available. Supplements were 0–10% fetal calf serum (FCS, Gibco) depending on the experiment and 1% antibiotic mixture (Penicillin-Streptomycin-Glutamine; Life Technologies). Transfection of DYNC2LI1 siRNA (HSS147516; Life Technologies) was carried out in medium without antibiotics using Lipofectamin 2000 (Life Technologies) according to the manufacturer’s instructions. Ciliogenesis was induced by incubating cells in serum-free medium (0% FCS) for a period of 5 days.
DYNC2LI1 knockdown
Stealth RNAi™ siRNA Negative Control and 3 DYNC2LI1 Stealth RNAi™ siRNAs (HSS182153, HSS147514, HSS147516) were purchased from Life Technologies. The DYNC2LI1 siRNA HSS147516 with the lowest level of residual activity in control experiments and the corresponding Stealth RNAi™ siRNA Negative Control Lo GC (scrambled control 12935-200) were used.
Immunofluorescence
For immunofluorescence analyses cells were fixed with an EGTA-saturated ice-cold 100% methanol solution. Cells were treated with 0.01% Tween for 10 min followed by blocking of non-specific protein interactions for 60 min with blocking solution (0.5% cold-water fish gelatin and 0.1% BSA in PBS). Incubation with primary antibodies (diluted in blocking solution) was performed overnight at 4° C. Centrosomes or the basal body of the primary cilium were stained with MmPeriC1 (rabbit, anti-Pcnt pAb, 1:500,79) and the primary cilium was stained with GT335 (mouse, anti-Polyglutamylation Modification mAb, 1:1000, AdipoGen, San Diego CA, USA). For DYNC2LI1 localization studies we used anti-DYNC2LI1 (rabbit, 15949-1-AP, 1:100, Acris, Herford, Germany). IFT57 (rabbit, 11083-1-AP, 1:100, Proteintech, Manchester, United Kingdom) and IFT88 (rabbit, 13967-1-AP, 1:200, Proteintech, Manchester, United Kingdom) antibodies were used for accumulations studies of IFT-B proteins in ciliary tips. Alexa Fluor® 488 goat anti- rabbit IgG (green, 1:500) and Alexa Fluor® 594 goat anti-mouse IgG (red, 1:500) (Life Technologies) were used as secondary antibodies and cell nuclei were stained with DAPI (4′,6-Diamidin-2-phenylindol, 1:50000, Serva, Heidelberg, Germany). Samples were mounted in Aqua Poly Mount (Polysciences, Eppelheim, Germany) and analyzed with a Zeiss Axio Imager Z2 fluorescence microscope equipped with an Apotome. Projections of z-stacks were calculated with AxioVision 4.8 software (Zeiss, Oberkochen, Germany) and the images were adjusted for brightness and contrast using Adobe Photoshop CS (Adobe, San Jose, CA). The length of primary cilia was measured with the FIJI program80.
We performed three-dimensional reconstruction of cilia using Imaris software 7.7 (Bitplane, Zurich, Switzerland) with picture stacks from a Laser Scanning Microscope 710 (Carl Zeiss) and ZEN 2010 software with corresponding imaging modules.
Additional Information
How to cite this article: Kessler, K. et al. DYNC2LI1 mutations broaden the clinical spectrum of dynein-2 defects. Sci. Rep. 5, 11649; doi: 10.1038/srep11649 (2015).
References
Fry, A. M., Leaper, M. J. & Bayliss, R. The primary cilium: guardian of organ development and homeostasis. Organogenesis 10, 62–68 (2014).
Goetz, S. C. & Anderson, K. V. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet 11, 331–344 (2010).
Satir, P., Pedersen, L. B. & Christensen, S. T. The primary cilium at a glance. J Cell Sci 123, 499–503 (2010).
Satir, P. & Christensen, S. T. Overview of structure and function of mammalian cilia. Annu Rev Physiol 69, 377–400 (2007).
van Reeuwijk, J., Arts, H. H. & Roepman, R. Scrutinizing ciliopathies by unraveling ciliary interaction networks. Hum Mol Genet 20, R149–R157 (2011).
Sasai, N. & Briscoe, J. Primary cilia and graded Sonic Hedgehog signaling. Wiley interdisciplinary reviews. Developmental biology 1, 753–772 (2012).
Oh, E. C. & Katsanis, N. Context-dependent regulation of Wnt signaling through the primary cilium. J Am Soc Nephrol 24, 10–18 (2013).
Sebbagh, M. & Borg, J. P. Insight into planar cell polarity. Exp Cell Res 328, 284–295 (2014).
Neugebauer, J. M., Amack, J. D., Peterson, A. G., Bisgrove, B. W. & Yost, H. J. FGF signalling during embryo development regulates cilia length in diverse epithelia. Nature 458, 651–654 (2009).
Leitch, C. C., Lodh, S., Prieto-Echague, V., Badano, J. L. & Zaghloul, N. A. Basal body proteins regulate Notch signaling through endosomal trafficking. J Cell Sci 127, 2407–2419 (2014).
Boehlke, C. et al. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat Cell Biol 12, 1115–1122 (2010).
Schneider, L. et al. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr Biol 15, 1861–1866 (2005).
Kim, M., Kim, M., Lee, M. S., Kim, C. H. & Lim, D. S. The MST1/2-SAV1 complex of the Hippo pathway promotes ciliogenesis. Nat Commun 5, 5370 (2014).
Gerdes, J. M., Davis, E. E. & Katsanis, N. The vertebrate primary cilium in development, homeostasis and disease. Cell 137, 32–45 (2009).
Eggenschwiler, J. T. & Anderson, K. V. Cilia and developmental signaling. Annu Rev Cell Dev Biol 23, 345–373 (2007).
Ishikawa, H. & Marshall, W. F. Ciliogenesis: building the cell’s antenna. Nat Rev Mol Cell Biol 12, 222–234 (2011).
Pedersen, L. B., Veland, I. R., Schroder, J. M. & Christensen, S. T. Assembly of primary cilia. Dev Dyn 237, 1993–2006 (2008).
Czarnecki, P. G. & Shah, J. V. The ciliary transition zone: from morphology and molecules to medicine. Trends Cell Biol 22, 201–210 (2012).
Reiter, J. F., Blacque, O. E. & Leroux, M. R. The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep 13, 608–618 (2012).
Pedersen, L. B., Schroder, J. M., Satir, P. & Christensen, S. T. The ciliary cytoskeleton. Comprehensive Physiology 2, 779–803 (2012).
Santos, N. & Reiter, J. F. Building it up and taking it down: the regulation of vertebrate ciliogenesis. Dev Dyn 237, 1972–1981 (2008).
Pedersen, L. B. & Rosenbaum, J. L. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr Top Dev Biol 85, 23–61 (2008).
Avasthi, P. & Marshall, W. F. Stages of ciliogenesis and regulation of ciliary length. Differentiation; research in biological diversity 83, S30–S42 (2012).
D’Angelo, A. & Franco, B. The dynamic cilium in human diseases. Pathogenetics 2, 3 (2009).
Huber, C. & Cormier-Daire, V. Ciliary disorder of the skeleton. Am J Med Genet C Semin Med Genet 160c, 165–174 (2012).
Schmidts, M. et al. Combined NGS approaches identify mutations in the intraflagellar transport gene IFT140 in skeletal ciliopathies with early progressive kidney Disease. Hum Mutat 34, 714–724 (2013).
Halbritter, J. et al. Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am J Hum Genet 93, 915–925 (2013).
Huber, C. et al. WDR34 mutations that cause short-rib polydactyly syndrome type III/severe asphyxiating thoracic dysplasia reveal a role for the NF-kappaB pathway in cilia. Am J Hum Genet 93, 926–931 (2013).
Schmidts, M. et al. Mutations in the gene encoding IFT dynein complex component WDR34 cause Jeune asphyxiating thoracic dystrophy. Am J Hum Genet 93, 932–944 (2013).
McInerney-Leo, A. M. et al. Short-rib polydactyly and Jeune syndromes are caused by mutations in WDR60. Am J Hum Genet 93, 515–523 (2013).
Bergmann, C. Educational paper: ciliopathies. Eur J Pediatr 171, 1285–1300 (2012).
Warman, M. L. et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A 155A, 943–968 (2011).
Panda, A., Gamanagatti, S., Jana, M. & Gupta, A. K. Skeletal dysplasias: A radiographic approach and review of common non-lethal skeletal dysplasias. World journal of radiology 6, 808–825 (2014).
Asante, D., Stevenson, N. L. & Stephens, D. J. Subunit composition of the human cytoplasmic dynein-2 complex. J Cell Sci 127, 4774–4787 (2014).
Pfister, K. K. et al. Genetic analysis of the cytoplasmic dynein subunit families. PLoS Genet 2, e1 (2006).
Abou Jamra, R. et al. Adaptor protein complex 4 deficiency causes severe autosomal-recessive intellectual disability, progressive spastic paraplegia, shy character and short stature. American journal of human genetics 88, 788–795 (2011).
Abou Jamra, R. et al. Homozygosity mapping in 64 Syrian consanguineous families with non-specific intellectual disability reveals 11 novel loci and high heterogeneity. European journal of human genetics : EJHG 19, 1161–1166 (2011).
Hoischen, A. et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat Genet 42, 483– 485
Krawitz, P. M. et al. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet 42, 827–829 (2010).
Ng, S. B. et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet 42, 790–793 (2010).
Zahnleiter, D. et al. MAP4-dependent regulation of microtubule formation affects centrosome, cilia and golgi architecture as a central mechanism in growth regulation. Hum Mutat 36, 87–97 (2015).
Leipe, D. D., Koonin, E. V. & Aravind, L. STAND, a class of P-loop NTPases including animal and plant regulators of programmed cell death: multiple, complex domain architectures, unusual phyletic patterns and evolution by horizontal gene transfer. J Mol Biol 343, 1–28 (2004).
Pampliega, O. et al. Functional interaction between autophagy and ciliogenesis. Nature 502, 194–200 (2013).
Valente, E. M., Rosti, R. O., Gibbs, E. & Gleeson, J. G. Primary cilia in neurodevelopmental disorders. Nature reviews. Neurology 10, 27–36 (2014).
Yang, S. S., Roth, J. A. & Langer, L. O., Jr. Short rib syndrome Beemer-Langer type with polydactyly: a multiple congenital anomalies syndrome. Am J Med Genet 39, 243–246 (1991).
Elcioglu, N. H. & Hall, C. M. Diagnostic dilemmas in the short rib-polydactyly syndrome group. Am J Med Genet 111, 392–400 (2002).
Jeune, M., Beraud, C. & Carron, R. [Asphyxiating thoracic dystrophy with familial characteristics]. Archives francaises de pediatrie 12, 886–891 (1955).
Baujat, G. & Le Merrer, M. Ellis-van Creveld syndrome. Orphanet J Rare Dis 2, 27 (2007).
van Dam, T. J. et al. The SYSCILIA gold standard (SCGSv1) of known ciliary components and its applications within a systems biology consortium. Cilia 2, 7 (2013).
Davis, E. E. & Katsanis, N. The ciliopathies: a transitional model into systems biology of human genetic disease. Curr Opin Genet Dev 22, 290–303 (2012).
Geremek, M. et al. Gene expression studies in cells from primary ciliary dyskinesia patients identify 208 potential ciliary genes. Hum Genet 129, 283–293 (2011).
Inglis, P. N., Boroevich, K. A. & Leroux, M. R. Piecing together a ciliome. Trends Genet 22, 491–500 (2006).
Rana, A. A. et al. Targeted deletion of the novel cytoplasmic dynein mD2LIC disrupts the embryonic organiser, formation of the body axes and specification of ventral cell fates. Development 131, 4999–5007 (2004).
Grissom, P. M., Vaisberg, E. A. & McIntosh, J. R. Identification of a novel light intermediate chain (D2LIC) for mammalian cytoplasmic dynein 2. Mol Biol Cell 13, 817–829 (2002).
Krock, B. L., Mills-Henry, I. & Perkins, B. D. Retrograde intraflagellar transport by cytoplasmic dynein-2 is required for outer segment extension in vertebrate photoreceptors but not arrestin translocation. Investigative ophthalmology & visual science 50, 5463–5471 (2009).
Perrone, C. A. et al. A novel dynein light intermediate chain colocalizes with the retrograde motor for intraflagellar transport at sites of axoneme assembly in chlamydomonas and Mammalian cells. Mol Biol Cell 14, 2041–2056 (2003).
Mikami, A. et al. Molecular structure of cytoplasmic dynein 2 and its distribution in neuronal and ciliated cells. J Cell Sci 115, 4801–4808 (2002).
Madhivanan, K. & Aguilar, R. C. Ciliopathies: the trafficking connection. Traffic 15, 1031–1056 (2014).
Merrill, A. E. et al. Ciliary abnormalities due to defects in the retrograde transport protein DYNC2H1 in short-rib polydactyly syndrome. Am J Hum Genet 84, 542–549 (2009).
May, S. R. et al. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev Biol 287, 378–389 (2005).
Schmidts, M. et al. Exome sequencing identifies DYNC2H1 mutations as a common cause of asphyxiating thoracic dystrophy (Jeune syndrome) without major polydactyly, renal or retinal involvement. J Med Genet 50, 309–323 (2013).
St-Jacques, B., Hammerschmidt, M. & McMahon, A. P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev 13, 2072–2086 (1999).
Anderson, E., Peluso, S., Lettice, L. A. & Hill, R. E. Human limb abnormalities caused by disruption of hedgehog signaling. Trends Genet 28, 364–373 (2012).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43, 491–498 (2011).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research 38, e164 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols 4, 1073–1081 (2009).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat Methods 7, 248–249 (2010).
Schwarz, J. M., Rodelsperger, C., Schuelke, M. & Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 7, 575–576 (2010).
Genomes Project, C. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012).
Robinson, J. T. et al. Integrative genomics viewer. Nat Biotechnol 29, 24–26 (2011).
Thorvaldsdottir, H., Robinson, J. T. & Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in bioinformatics 14, 178–192 (2013).
McDowall, J. & Hunter, S. InterPro protein classification. Methods Mol Biol 694, 37–47 (2011).
Kurowski, M. A. & Bujnicki, J. M. GeneSilico protein structure prediction meta-server. Nucleic Acids Res 31, 3305–3307 (2003).
Webb, B. & Sali, A. Protein structure modeling with MODELLER. Methods Mol Biol 1137, 1–15 (2014).
Guex, N. & Peitsch, M. C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723 (1997).
Sayle, R. A. & Milner-White, E. J. RASMOL: biomolecular graphics for all. Trends in biochemical sciences 20, 374 (1995).
Muhlhans, J., Brandstatter, J. H. & Giessl, A. The centrosomal protein pericentrin identified at the basal body complex of the connecting cilium in mouse photoreceptors. PLoS One 6, e26496 (2011).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682 (2012).
Acknowledgements
We thank the patients and their family for their kind cooperation and Evelyn Galsterer for her excellent technical assistance. This study was supported by the DFG grant TH 896/3-3 to C. Thiel and by the IZKF (Interdisciplinary Centre for Clinical Research of the Universität of Erlangen-Nürnberg) Grant F4 and approved by the Ethical Review Board of the Medical Faculty of the Friedrich-Alexander-Universität Erlangen-Nürnberg and the families gave their consent. The authors would like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about.
Author information
Authors and Affiliations
Contributions
K.K., I.W. and N.S.F. performed experiments, A.G., A.R., R.R., J.H.B. and C.T.T. supervised the experiments and corrected the manuscript, S.U., B.P. and A.B.E. performed and analyzed the exome sequencing, R.R. and A.G. supplied antibodies, H.S. did protein modeling analysis, E.S. supplied patient data and material, P.K. supplied material, H.-G.D. provided clinical expertise, K.K. and C.T.T. wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kessler, K., Wunderlich, I., Uebe, S. et al. DYNC2LI1 mutations broaden the clinical spectrum of dynein-2 defects. Sci Rep 5, 11649 (2015). https://doi.org/10.1038/srep11649
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep11649
This article is cited by
-
Combinations of deletion and missense variations of the dynein-2 DYNC2LI1 subunit found in skeletal ciliopathies cause ciliary defects
Scientific Reports (2022)
-
Potassium Channel KCNH1 Activating Variants Cause Altered Functional and Morphological Ciliogenesis
Molecular Neurobiology (2022)
-
Dynein tails: how to hitch a ride on an IFT train
Nature Structural & Molecular Biology (2019)
-
WDR34 Activates Wnt/Beta-Catenin Signaling in Hepatocellular Carcinoma
Digestive Diseases and Sciences (2019)
-
Functional missense and splicing variants in the retinoic acid catabolizing enzyme CYP26C1 in idiopathic short stature
European Journal of Human Genetics (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
