
Abstract
The underlying cause of the multiple congenital anomalies/mental retardation syndrome Kabuki syndrome (KS, OMIM 147920) has not yet been established. We identified seven patients who fulfilled the classical clinical criteria for this syndrome and undertook a detailed clinical, ophthalomological and molecular cytogenetic review. Three of the seven patients had previously undetected ocular anomalies including myopia, ptosis, strabismus and tilted discs. The identification of preventable causes of loss of vision underlines the value of detailed ophthalmologic examination of KS patients. Using BAC fluorescence in situ hybridisation, there was no evidence of the duplication of 8p recently reported by Milunsky and Huang. We conclude that the cause of KS has yet to be established.
Similar content being viewed by others


Introduction
Kabuki syndrome (KS, OMIM 147920) is a multiple congenital anomalies/mental retardation syndrome of unknown cause. It is characterised by mild to moderate mental retardation, postnatal growth retardation, skeletal anomalies and classical facial features.1, 2 The facial gestalt is important in making the diagnosis. In particular, the eye findings of long palpebral fissures and eversion of the lateral third of the lower eyelid are suggestive. Recent reports have noted a high incidence of ocular abnormalities in this group of patients.3, 4
Several inconsistent chromosome aberrations have been described in KS patients of which microdeletions of 1q32-41,5 22q11.26 and ring X chromosomes7 have already been excluded. Recently, high-resolution comparative genomic hybridisation (HR-CGH) was used to detect a duplication of 8p22 to 8p23.1 in 6/6 patients with KS8 and a circa 3.5 megabase duplication was identified in all patients using locus-specific FISH probes. We set out to review a series of clinically well-defined patients with KS to characterise their eye findings and to replicate the findings of Milunsky and Huang.8
Materials and methods
Patient identification
Patients with a clinical diagnosis of KS as agreed by four consultant clinical geneticists were identified from the Wessex Clinical Genetics Service Database.
Cytogenetic and molecular cytogenetic methods
High-resolution G-banded analysis at the 550+ band level and fluorescence in situ hybridisation (FISH) with two of the same probes used by Milunsky and Huang8 were performed using standard techniques. RP11-112G9 maps to medial 8p23.1 and RP11-92C1 maps to distal 8p22. Two independent observers scored 10 metaphases and 10 interphases for each probe. Both probes were kindly provided by the Sanger Centre, Hinxton, Cambridge, UK from the Ensembl 1 Megabase cloneset (www.ensembl.org/homo_sapiens/cytoview).
Results
Seven patients (five female and two male subjects) with KS attended a genetic and ophthalmology clinic. They underwent full clinical examination including formal ophthalmology and orthoptic review. Five patients agreed to repeat blood sampling, but all seven patients had previously been karyotyped.
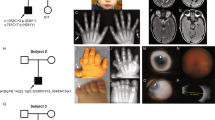
The phenotypic features of each of the patients are detailed in Table 1, the ocular findings in Table 2. Photographs of four of the patients are shown in Figure 1.

Patient photographs.
Cytogenetics and molecular cytogenetics
We expected that high-resolution chromosome analysis directed at medial 8p would reveal cytogenetically visible duplications because the estimated size of the duplications was 3.5 Mb which is within the 2–4 Mb resolution of the light microscope. However, no cytogenetic evidence of a duplication of 8p was found and all 7/7 patients were cytogenetically normal at the 550 band level. In 5/7 patients, this was confirmed using FISH with the same probes that defined the medial 8p23.1 (112G9) and distal 8p22 (92C1) boundaries of the duplications reported by Milunsky and Huang.8 In each patient, the probes hybridised to 8p in the expected locations with no evidence of duplication in either metaphases or interphase nuclei (Figure 2). The karyotypes were therefore: 46,XX or XY.ish 8p23.1(112G9x2), 8p22(92C1x2).

(a) Representative FISH metaphase and (b) interphases using probe 112G9 (red) and the 8 centromere (green).
Discussion
KS is a multisystem disorder and our cohort is typical with evidence of abnormal development of the face, eyes, skeleton, heart, kidney and developmental delay. The presence of a bicuspid aortic valve and coarctation in two of five patients is noteworthy as this was found to be the most common type of congenital heart disease in KS patients.9 There were individual patients with congenital pseudarthrosis of the clavicle and immunodeficiency. Disorders in a similar spectrum to each of these have been previously reported in KS patients.10, 11
The external eye and eyebrow findings are important diagnostic criteria1 and are certainly important features of the gestalt. Our review of seven KS patients showed that they all have palpebral fissure lengths greater than two standard deviations from the mean when plotted for age. This was most significant at younger ages. Thus, formal palpebral fissure measurement could be useful diagnostically.
During this review, newly diagnosed ocular findings in three patients included one with a previously unknown refractive error (myopia), one with strabismus and tilted discs and one with a previously unrecognised ptosis. This supports the recommendations that all patients with a diagnosis of KS should have a formal ophthalmology review.3, 4 It is particularly important to identify and correct potentially reversible causes of reduced vision in developmentally delayed patients. This group may not communicate their visual difficulties, yet visual defect correction can help minimise any negative impact on development. There are a multitude of other ophthalmologic findings reported in KS.3, 4
Nocturnal lagophthalmos (sleeping with eyes open) has also been reported to occur in over half of KS patients.12 This can predispose to eye complications such as keratitis or conjunctivitis. Discussion with the parents of our patients confirmed that nocturnal lagophthalmos is common, occurring in four out of five on direct questioning.
In our clinically well-defined cohort, there was no conventional or molecular cytogenetic evidence of the duplication of 8p22–p23.1 that has been reported.8 There are several possible explanations.
Firstly, KS may be genetically heterogeneous with some patients having a duplication and others an as yet unidentified genetic cause. Our cohort may thus represent a different genetic subset. Given the various karyotypic anomalies previously described, this remains a distinct possibility.
Secondly, it is possible that the diagnosis is inaccurate in some of our cohort or some of the patients studied by Milunsky and Huang.8 Interpreting reproduced photographs can be difficult especially if patients are from different racial groups, but the photographs published by Milunsky and Huang5 do not demonstrate some of the facial features normally considered classical for KS. For example, the photographs do not show high arched eyebrows or prominent outstanding ears.
The third and final possibility for not finding the same duplication of 8p22–23.1 in this study could be for technical reasons. This seems unlikely as high-resolution cytogenetic analysis and FISH, using the same probes as Milunsky and Huang in 5/7 patients, were both normal and concordant. However, our results cannot exclude the possibility of a subcytogenetic or molecular duplication within this region.
Our results support the need for careful ophthalmalogical evaluation of KS patients but, like those of Miyake et al,13, 14, 15 are not consistent with the findings of Milunsky and Huang.8 We conclude that the cause of KS remains to be established.
References
Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, Kajji T : Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J Pediatr 1981; 99: 565–569.
Niikawa N, Kuroki Y, Kajii T et al: Kabuki make-up (Niikawa–Kuroki) syndrome: a study of 62 patients. Am J Med Genet 1988; 31: 565–590.
Klujit I, van Dorp DB, Kwee ML et al: Kabuki syndrome – report of six cases and review literature with emphasis on ocular features. Ophthalm Genet 2000; 21: 51–61.
Ming JE, Russell KL, Bason L, McDonald-McGinn DM, Zackai EH : Coloboma and other ophthalmologic anomalies in kabuki syndrome: distinct from Charge association. Am J Med Genet 2003; 123A: 249–252.
Makita Y, Yamada K, Miyamoto A, Okuno A, Niikawa N : Kabuki make-up syndrome is not caused by microdeletion close to the van der Woude syndrome critical region at 1q32–q41. Am J Med Genet 1999; 86: 285–288.
Li M, Zackai EH, Niikawa N, Kaplan P, Driscoll DA : Kabuki syndrome is not caused by a microdeletion in the DiGeorge/velocardiofacial chromosomal region within 22q11.2. Am J Med Genet 1996; 65: 101–103.
Leppig KA, Sybert VP, Ross JL et al: Phenotype and X inactivation in 45,X/46,X,r(X) cases. Am J Med Genet 2004; 128a: 276–284.
Milunsky JM, Huang XL : Unmasking Kabuki syndrome: chromosome 8p22–8p23.1 duplication revealed by comparative genomic hybridisation and BAC-FISH. Clin Genet 2003; 64: 509–516.
Digilio M, Marino B, Toscano A, Giannotti A, Dallapiccola B : Congenital heart defects in Kabuki syndrome. Am J Med Genet 2001; 100: 269–274.
Fryns JP, Devriendt K : Hypoplastic claviculae in the kabuki (Niikawa–Kuroki) syndrome. Genet Couns 1998; 9: 57–58, (letter).
Chrzanowska KH, Krajewska-Walasek M, Kus J et al: Kabuki (Niikawa–Kuroki) syndrome associated with immunodeficiency. Clin Genet 1998; 53: 308–312.
Toriello HV, Droste P : Nocturnal lagophthalmos in Kabuki syndrome: results of a parent group survey. Proc Greenwood Genet Cent 2003; 22: 110, (abstract).
Miyake N, Harada N, Shimokawa O et al: On the reported 8p22–p23.1 duplication in Kabuki make-up syndrome (KMS) and its absence in patients with typical KMS. Am J Med Genet 2004; 128A: 170–172.
Engelen JJ, Loneus WH, Vaes-Poeters G, Schrander-Stumpel CT : Kabuki syndrome is not caused by an 8p duplication: a cytogenetic study in 20 patients. Am J Med Genet A 2005; 132: 276–277.
Hoffman JD, Zhang Y, Greshock J et al: Array based CGH and FISH fail to confirm duplication of 8p22–p23.1 in association with Kabuki syndrome. J Med Genet 2005; 42: 49–53.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Turner, C., Lachlan, K., Amerasinghe, N. et al. Kabuki syndrome: new ocular findings but no evidence of 8p22-p23.1 duplications in a clinically defined cohort. Eur J Hum Genet 13, 716–720 (2005). https://doi.org/10.1038/sj.ejhg.5201377
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201377
Keywords
This article is cited by
-
Ocular Involvement in Primary Immunodeficiency Diseases
Journal of Clinical Immunology (2014)
-
New fundus findings in a case of Kabuki syndrome
Eye (2009)
-
BAC-FISH refutes report of an 8p22–8p23.1 inversion or duplication in 8 patients with Kabuki syndrome
BMC Medical Genetics (2006)
