
Abstract
Recently, variants in CHEK2 gene were shown to associate with sporadic prostate cancer in the USA. In the present study from Finland, we found that the frequency of 1100delC, a truncating variant that abrogates the kinase activity, was significantly elevated among 120 patients with hereditary prostate cancer (HPC) (four out of 120 (3.3%); odds ratio 8.24; 95% confidence interval 1.49–45.54; P=0.02) compared to 480 population controls. Suggestive evidence of segregation between the 1100delC mutation and prostate cancer was seen in all positive families. In addition, I157T variant had significantly higher frequency among HPC patients (13 out of 120 (10.8%); odds ratio 2.12; 95% confidence interval 1.06–4.27; P=0.04) than the frequency 5.4% seen in the population controls. The results suggest that CHEK2 variants are low-penetrance prostate cancer predisposition alleles that contribute significantly to familial clustering of prostate cancer at the population level.
Similar content being viewed by others
Main
Analyses using families with hereditary prostate cancer (HPC) have suggested that multiple genetic loci may harbour prostate cancer susceptibility genes, including HPC1 (MIM 601518) at 1q24–q25, HPC2 (MIM 605367) at 17p11, PCAP (MIM 602759) at 1q42–q43, HPCX (MIM 300147) at Xq27–q28, CAPB (MIM 603688) at 1p36, and HPC20 (MIM 176807) at 20q13 (Nwosu et al, 2001). So far, only two genes have been identified from these chromosomal regions: ELAC2 from HPC2 –locus (Tavtigian et al, 2001) and RNASEL (MIM 180435) from HPC1 –locus (Carpten et al, 2002). In Finland neither ELAC2 nor RNASEL did explain the disease segregation in HPC families, but seemed to have some kind of modifying role in prostate carcinogenesis (Rokman et al, 2001; Rokman et al, 2002). Xu et al (2001) identified a new locus at 8p22–23, and mutations in MSR1 gene (MIM 153622) were reported to associate with prostate cancer (Xu et al, 2002). However, recent results do not support a major role for the MSR1 gene in the causation of prostate cancer (Seppala et al, in press). Definitive confirmations of the role of ELAC2, RNASEL, or MSR1 in prostate cancer predisposition are still warranted.
Recently, mutations in CHEK2 (MIM 604373) were identified in patients with prostate cancer (Dong et al, 2003). The CHEK2 gene localises to chromosome 22q12.1 and contains 14 exons. Originally, germline mutations in CHEK2 gene were reported in Li–Fraumeni syndrome and breast cancer (Bell et al, 1999; Allinen et al, 2001). Rare somatic mutations in CHEK2 have also been identified in a number of cancer types, including lung and ovarian cancers and osteosarcomas (Miller et al, 2002). These results together with the normal function of CHEK2 in DNA damage checkpoints are consistent with the idea that CHEK2 might act as a tumour suppressor gene. Here, we explored the significance of CHEK2 gene in prostate cancer causation in Finland. The Finnish population is known to be historically isolated and genetically homogeneous (Peltonen et al, 2000). Therefore, there may be a limited number of prostate cancer causing mutations, and the effect of individual risk genes could be identified more readily than in more heterogeneous populations.
Materials and methods
Families with HPC
Collection of Finnish families with HPC has been described elsewhere (Schleutker et al, 2000). For single-strand conformation polymorphism (SSCP) analysis, youngest affected patient from each of the 120 HPC families was initially used for the screening of CHEK2 mutations. The HPC families consisted of two cohorts. The families in the first cohort (n=68) had either three or more first- or second-degree- affected members or two affected members with at least the index patient diagnosed with prostate cancer ⩽60 years of age. The mean age at diagnosis for the index patients was 62.2 years (range 44–81 years), and the mean number of affected family members was 3.2 (range 2–6). The second cohort of families (n=52) had only two first- or second-degree affected members with ages at diagnosis >60 years. The mean age at diagnosis for the index patients was 69.0 years (range 61–86).
Patients with prostate cancer and controls
The 1100delC and I157T variants were analysed in 537 patients with unselected prostate cancer, and in 480 healthy male blood donors. There were altogether 634 consecutive patients diagnosed with prostate cancer in the Pirkanmaa Hospital District with a population of around 450 000 during 1999–2000. We had samples from 85% of these patients, which results in an unselected, population-based collection of patients. The mean age at diagnosis for the patients with unselected prostate cancer was 68.6 years (range 47–90). Information was available on the tumour WHO-grade in 96%, on Gleason score in 88%, on T-stage in 100%, and on N-stage in 30% of the patients. M-stage was ascertained by bone scan in 73% of the patients with PSA⩾10 μg l−1 and in 26% of the patients with PSA<10 μg l−1. In all, 12% (66 out of 537) of the patients reported a positive family history of prostate cancer. The controls consisted of DNA samples from anonymous male blood donors obtained from the Blood Center of the Finnish Red Cross in Tampere.
Written informed consent was obtained from all living patients and also, for families with HPC, from the unaffected members. The research protocols were approved by the Ethical Committee of the Tampere University Hospital (93175, 95062, and 99228), and the National Human Genome Research Institute (HG-0158). Permission for collection of families, in the entirety of Finland, was granted by the Ministry of Health and Social Affairs (59/08/95).
Mutation screening with SSCP analysis
Single-strand conformation polymorphism analysis of the entire coding sequence of the CHEK2 gene was designed to include all intron–exon boundaries (GenBank accession number AF086904). Primers used for amplification of exons 10–14, which are known to be repeated on several other chromosomes (Sodha et al, 2000), were designed so that both primers for each primer pair had a base mismatch in the most 3′ nucleotide, compared with sequences from nonfunctional copies of CHEK2. Genomic DNA was used at 25 ng per 15 μl reaction mixture containing 1.5 mM MgCl2; 20 μ M each of dNTP; 0.5 μCi of α(33P)-dCTP (Amersham Pharmacia, Uppsala, Sweden); 0.6 μ M of each primer; 1.0 U AmpliTaqGold; and the reaction buffer provided by the supplier (PE Biosystems, Foster City, CA, USA). Annealing temperature of 50°C (for exons 10 and 12) or 55°C (for all other exons) was used. After denaturation, the (33P)-labeled PCR products were electrophoresed at 800 V for 12 h at room temperature, in 0.5 × mutation-detection-enhancement gel (FMC BioProducts, Rockland, ME, USA) with 1% glycerol in 0.5 × Tris-borate EDTA. For exon 11, the electrophoresis was also performed without glycerol in the gel. After electrophoresis, gels were dried and exposed to Kodak BioMax maximum-resolution films for 6 h. All samples, in which variant bands were detected, as well as two normal bands per exon, were sequenced using the same PCR primers and ABI Prism 310 Genetic Analyzer (PE Biosystems, Foster City, CA, USA). Also, the genotypes of the available family members of the mutation carriers were determined by sequencing.
Minisequencing and SSCP for large-scale population screening of identified variants
The frequencies of the two CHEK2 variants were determined in the entire sample of patients described above. 1100delC variant was screened by minisequencing (Syvanen, 1998). PCR was performed with 100 ng of DNA, 0.2 μ M each primer, 0.2 mM each dNTP, 1.5 mM MgCl2, and 1.0 U of AmpliTaqGold (PE Biosystems, Foster City, CA, USA), in a final volume of 50 μl. I157T variant was screened by SSCP analysis as described above. Positive results from both mutation analyses were confirmed by sequencing.
Statistical analyses
Association of the CHEK2 genotypes with HPC and unselected prostate cancer was tested by logistic-regression analysis, by use of the SPSS statistical software package (SPSS 11.0). Association with demographic, clinical, and pathological features of the disease was tested by the Mann–Whitney test, Pearson χ2 test, and Fisher's exact test by use of the SPSS statistical software package (SPSS 11.0).
Results
Five sequence variants were identified in the SSCP analysis of the CHEK2 gene in 120 index patients from Finnish families with HPC (Table 1). Two of the variants, a missense variant 470T>C (I157T) in exon 3 and a frameshift mutation 1100delC in exon 10, were the same as previously reported in patients with Li–Fraumeni syndrome (Bell et al, 1999), breast (Allinen et al, 2001; Vahteristo et al, 2002), and prostate cancer (Dong et al, 2003). The frameshift 1100delC mutation has been proven to result in the loss of kinase activity (Wu et al, 2001), and I157T variant has been shown to be defective in its ability to bind and phosphorylate Cdc25A, one of its normal substrates (Falck et al, 2001). These variants were further studied in a set of 1137 samples. In addition, a silent exonic change also reported by Bell et al (1999), an intronic change (not affecting splice site), and a novel missense mutation 1312G>T (D438Y) were observed. D438Y mutation was found only in one proband. In this family there were two prostate cancer patients. Unfortunately, we did not have a sample from the second affected person (deceased).
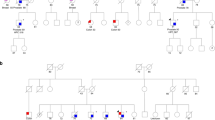
The 120 patients with HPC included 3.3% (four out of 120) patients who carried the 1100delC mutation (Table 2). This was significantly higher (odds ratio (OR)=8.24; 95% confidence interval (CI) 1.49−45.54; P=0.02) than the frequency 0.4% seen in population sample of 480 blood donors. Among the unselected patients with prostate cancer, the frequency of 1100delC variant was 1.3% (seven out of 537). All 1100delC carriers were heterozygous. All other affected and unaffected male relatives were also genotyped from 1100delC-positive families. Suggestive evidence of segregation between the 1100delC mutation and prostate cancer was seen in all four families (Figure 1). Since 1100delC mutation has been reported in Li–Fraumeni syndrome and was previously called a mutation hot spot (CHEK2 [MIM 604373]), we looked other cancers in these four 1100delC-positive families. In family 001, there was one second-degree relative diagnosed with lung cancer, but no other cancers were found among first- or second-degree relatives.

Segregation of CHEK2 1100delC mutation in four families with HPC. 1100delC variant carriers are denoted by a plus sign (+), and noncarriers by a minus sign (−). An asterisk (*) denotes the persons with no sample available. No sample was available from affected father (II-2) in family 351, but because the mother (II-1) did not carry the mutation, the father is a likely 1100delC mutation carrier. Current age of the unaffected members or age at diagnosis for prostate cancer patients (in years) is indicated below the symbol for each family member. In each family, the index patient is marked with an arrow. Squares denote male subjects, and circles denote female subjects; black symbols denote patients with prostate cancer.
The I157T variant was seen in 10.8% (13 out of 120) of patients with HPC (Table 2). This was also significantly higher (OR=2.12; 95% CI 1.06−4.27; P=0.04) than the frequency of 5.4% seen in the population controls. Nine of the I157T-positive families had only two affecteds, three families had three affecteds, and one family had four affecteds. Segregation of the variant with the disease was incomplete, in that both unaffected mutation carriers and mutation-negative patients with prostate cancer were observed. In addition, the I157T variant was found in 7.8% (42 out of 537) of unselected prostate cancer patients. One of these carriers was homozygous; all other I157T variant carriers were heterozygous. The homozygous carrier did not have family history of cancer and there was nothing unusual in his phenotype. None of the patients or controls carried both 1100delC and I157T variant.
The mean ages at diagnosis of the CHEK2 variant carriers in patients with HPC were 62.7 years for 1100delC carriers and 64.0 years for I157T carriers. These ages were only marginally different from the mean age of HPC patients with no mutations (65.2 years for both variants; P=0.57 for 1100delC and P=0.62 for I157T). A similar trend was observed in the cohort of unselected prostate cancer patients (64.6 vs 68.7 years; P=0.18 for 1100delC and 68.4 vs 68.6 years; P=0.86 for I157T). The association between the frequency of the two variants and disease phenotype, including tumour WHO-grade, Gleason score, T-, N- and M-stage and PSA value at diagnosis, were also analysed among unselected prostate cancer cases. No significant associations emerged from these analyses (data not shown).
Discussion
CHEK2 has been suggested to be a candidate tumour suppressor gene on the basis of the findings that normal function of CHEK2 is involved in DNA-damage respond and some of the mutations identified in Li–Fraumeni families were expected to result in a truncated protein (Bell et al, 1999). Subsequently, these findings were supported by the reports concerning identical and additional mutations in patients with Li–Fraumeni syndrome (Lee et al, 2001) and breast cancer (Meijers-Heijboer et al, 2002; Vahteristo et al, 2002). While this manuscript was in preparation, another study was published showing that mutations in CHEK2 were associated also with prostate cancer risk (Dong et al, 2003).
Our results suggest that CHEK2 1100delC mutation is associated with positive family history of prostate cancer. The mutation segregated almost completely in all mutation-positive families (Figure 1). In family 351, there were three unaffected men, who carried the variant. Two of them were rather young, about 50 years old (III-2 and III-3), and the third unaffected carrier was 71 years old (II-5). The total PSA values of the unaffected mutation carriers of this family were measured in July 2000. The values were <0.5, 2.2, and 2.4 μg l−1 for III-2, III-3 and II-5, respectively. The mean age at diagnosis of prostate cancer in Finland was 71.1 years in 1999 (Finnish Cancer Registry; cancer statistics at http://www.cancerregistry.fi/) and all three affecteds of the family 351 were over 66 years old when diagnosed for prostate cancer, thus the future diagnosis of prostate cancer cannot be ruled out for the healthy carriers of this family. On the other hand, in the four 1100delC-positive families, there were no mutation-negative prostate cancer patients. The association of 1100delC mutation with families that include small number of affected relatives, the most common types of prostate cancer families, implies that the mutation is likely to have a significant contribution to familial prostate cancer at the population level. In addition, I157T seems to be a disease-associated polymorphism at least in the Finnish population. It has a slightly higher frequency among patients with unselected prostate cancer than among control individuals and it is strongly associated with family history of the disease. However, according to the previous reports, the I157T allele does not make a significant contribution to breast cancer susceptibility (Allinen et al, 2001; Schutte et al, 2003). Therefore, the association with this allele is less conclusive.
Previously, Vahteristo et al (2002) reported the strong association of the CHEK2 1100delC with breast cancer families that included only two affected patients, suggesting that 1100delC is a low-penetrance genetic alteration. In contrast to our results, Dong et al (2003) reported the association of the CHEK2 mutations (all mutations pooled together) only with sporadic prostate cancer. In addition, they did not observe any association between prostate cancer and I157T variant. The reason why Dong et al (2003) did not detect any association with HPC could be due to different sample settings: the families from the USA represent more extreme HPC families than the Finnish families in the present study. In their study two affected members from 149 HPC families with at minimum of three affected men over at least two generations were used. In our recent genome-wide linkage analysis, no positive signals were seen on chromosome 22 (Schleutker et al, in press). This is probably due to the selected study material, as only the most extreme families were genotyped, possibly reflecting the same phenomenon as seen in the study of Dong et al (2003). Also, the low allele frequencies of CHEK2 variants (<10%) make this kind of an association almost impossible to detect by linkage analysis.
Dong et al (2003) reported a total of 13 different CHEK2 germline mutations among 400 sporadic prostate cancer patients and 298 individuals with familial prostate cancer. Most of these mutations occurred only once in their study population. The reason why fewer variants were found in our study can possibly be due to the limited sensitivity of the SSCP analysis and the number of screened patients. Most likely, however, the reason is the study population itself. The Finnish population is genetically much more homogeneous than the US population, and therefore it is not surprising that fewer variants were detected.
Taken together, finding of the 1100delC and I157T variants in families with small numbers of affected relatives support the idea that CHEK2 variants are low-penetrance prostate cancer predisposition alleles that contribute significantly to familial clustering of prostate cancer at the population level, especially in families with small number of affected relatives. However, variants in CHEK2 gene alone do not explain the familial clustering of prostate cancer in Finland as the majority of families did not have any CHEK2 alterations. The present results warrant further studies of the role of CHEK2 variants as a risk factor for prostate cancer in other populations.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Allinen M, Huusko P, Mantyniemi S, Launonen V, Winqvist R (2001) Mutation analysis of the CHK2 gene in families with hereditary breast cancer. Br J Cancer 85: 209–212
Bell DW, Varley JM, Szydlo TE, Kang DH, Wahrer DC, Shannon KE, Lubratovich M, Verselis SJ, Isselbacher KJ, Fraumeni JF, Birch JM, Li FP, Garber JE, Haber DA (1999) Heterozygous germ line hCHK2 mutations in Li–Fraumenisyndrome. Science 286: 2528–2531
Carpten J, Nupponen N, Isaacs S, Sood R, Robbins C, Xu J, Faruque M, Moses T, Ewing C, Gillanders E, Hu P, Bujnovszky P, Makalowska I, Baffoe-Bonnie A, Faith D, Smith J, Stephan D, Wiley K, Brownstein M, Gildea D, Kelly B, Jenkins R, Hostetter G, Matikainen M, Schleutker J, Klinger K, Connors T, Xiang Y, Wang Z, De Marzo A, Papadopoulos N, Kallioniemi OP, Burk R, Meyers D, Gronberg H, Meltzer P, Silverman R, Bailey-Wilson J, Walsh P, Isaacs W, Trent J (2002) Germline mutations in the ribonuclease L gene in families showing linkage with HPC1. Nat Genet 30: 181–184
Dong X, Wang L, Taniguchi K, Wang X, Cunningham JM, McDonnell SK, Qian C, Marks AF, Slager SL, Peterson BJ, Smith DI, Cheville JC, Blute ML, Jacobsen SJ, Schaid DJ, Tindall DJ, Thibodeau SN, Liu W (2003) Mutations in CHEK2 associated with prostate cancer risk. Am J Hum Genet 72: 270–280
Falck J, Mailand N, Syljuasen RG, Bartek J, Lukas J (2001) The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 410: 842–847
Lee SB, Kim SH, Bell DW, Wahrer DC, Schiripo TA, Jorczak MM, Sgroi DC, Garber JE, Li FP, Nichols KE, Varley JM, Godwin AK, Shannon KM, Harlow E, Haber DA (2001) Destabilization of CHK2 by a missense mutation associated with Li–Fraumeni Syndrome. Cancer Res 61: 8062–8067
Meijers-Heijboer H, van den Ouweland A, Klijn J, Wasielewski M, de Snoo A, Oldenburg R, Hollestelle A, Houben M, Crepin E, van Veghel-Plandsoen M, Elstrodt F, van Duijn C, Bartels C, Meijers C, Schutte M, McGuffog L, Thompson D, Easton D, Sodha N, Seal S, Barfoot R, Mangion J, Chang-Claude J, Eccles D, Eeles R, Evans DG, Houlston R, Murday V, Narod S, Peretz T, Peto J, Phelan C, Zhang HX, Szabo C, Devilee P, Goldgar D, Futreal PA, Nathanson KL, Weber B, Rahman N, Stratton MR (2002) Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet 31: 55–59
Miller CW, Ikezoe T, Krug U, Hofmann WK, Tavor S, Vegesna V, Tsukasaki K, Takeuchi S, Koeffler HP (2002) Mutations of the CHK2 gene are found in some osteosarcomas, but are rare in breast, lung, and ovarian tumors. Genes Chromosomes Cancer 33: 17–21
Nwosu V, Carpten J, Trent JM, Sheridan R (2001) Heterogeneity of genetic alterations in prostate cancer: evidence of the complex nature of the disease. Hum Mol Genet 10: 2313–2318
Peltonen L, Palotie A, Lange K (2000) Use of population isolates for mapping complex traits. Nat Rev Genet 1: 182–190
Rokman A, Ikonen T, Mononen N, Autio V, Matikainen MP, Koivisto PA, Tammela TL, Kallioniemi OP, Schleutker J (2001) ELAC2/HPC2 involvement in hereditary and sporadic prostate cancer. Cancer Res 61: 6038–6041
Rokman A, Ikonen T, Seppala EH, Nupponen N, Autio V, Mononen N, Bailey-Wilson J, Trent J, Carpten J, Matikainen MP, Koivisto PA, Tammela TL, Kallioniemi OP, Schleutker J (2002) Germline alterations of the RNASEL gene, a candidate HPC1 gene at 1q25, in patients and families with prostate cancer. Am J Hum Genet 70: 1299–1304
Schleutker J, Baffoe-Bonnie A, Gillanders E, Kainu T, Jones MP, Gildea D, Riedesel E, Albertus J, Freas-Lutz D, Markey C, Gibbs KD, Matikainen M, Koivisto P, Tammela T, Bailey-Wilson J, Trent J, Kallioniemi OP . A genome-wide scan for linkage in Finnish hereditary prostate cancer (HPC) families identifies novel susceptibility loci at 11q14 and 3p25–p26. Prostate, Unpublished manuscript listed in Human Relations Area Files (HRAF) eHRAF collection of ethnography: a world of cultures at your fingertips. http://ets.umdl.umich.edu/e/chrafe
Schleutker J, Matikainen M, Smith J, Koivisto P, Baffoe-Bonnie A, Kainu T, Gillanders E, Sankila R, Pukkala E, Carpten J, Stephan D, Tammela T, Brownstein M, Bailey-Wilson J, Trent J, Kallioniemi OP (2000) A genetic epidemiological study of hereditary prostate cancer (HPC) in Finland: frequent HPCX linkage in families with late-onset disease. Clin Cancer Res 6: 4810–4815
Schutte M, Seal S, Barfoot R, Meijers-Heijboer H, Wasielewski M, Evans DG, Eccles D, Meijers CFL, Klijn J, van den Ouweland A, Consortium TBCL, Futreal PA, Nathanson KL, Weber B, Easton D, Stratton MR, Rahman N (2003) Variants in CHEK2 other than 1100delC do not make a major contribution to breast cancer susceptibility. Am J Hum Genet 72: 1023–1028
Seppala EH, Ikonen T, Autio V, Rokman A, Mononen N, Matikainen M, Tammela T, Schleutker J . Germline alterations in MSR1 gene and prostate cancer risk. Clin Cancer Res, in press
Sodha N, Williams R, Mangion J, Bullock SL, Yuille MR, Eeles RA (2000) Screening hCHK2 for mutations. Science 289: 359
Syvanen AC (1998) Solid-phase minisequencing as a tool to detect DNA polymorphism. Methods Mol Biol 98: 291–298
Tavtigian SV, Simard J, Teng DH, Abtin V, Baumgard M, Beck A, Camp NJ, Carillo AR, Chen Y, Dayananth P, Desrochers M, Dumont M, Farnham JM, Frank D, Frye C, Ghaffari S, Gupte JS, Hu R, Iliev D, Janecki T, Kort EN, Laity KE, Leavitt A, Leblanc G, McArthur-Morrison J, Pederson A, Penn B, Peterson KT, Reid JE, Richards S, Schroeder M, Smith R, Snyder SC, Swedlund B, Swensen J, Thomas A, Tranchant M, Woodland AM, Labrie F, Skolnick MH, Neuhausen S, Rommens J, Cannon-Albright LA (2001) A candidate prostate cancer susceptibility gene at chromosome 17p. Nat Genet 27: 172–180
Vahteristo P, Bartkova J, Eerola H, Syrjakoski K, Ojala S, Kilpivaara O, Tamminen A, Kononen J, Aittomaki K, Heikkila P, Holli K, Blomqvist C, Bartek J, Kallioniemi OP, Nevanlinna H (2002) A CHEK2 genetic variant contributing to a substantial fraction of familial breast cancer. Am J Hum Genet 71: 432–438
Wu X, Webster SR, Chen J (2001) Characterization of tumor-associated Chk2 mutations. J Biol Chem 276: 2971–2974
Xu J, Zheng SL, Hawkins GA, Faith DA, Kelly B, Isaacs SD, Wiley KE, Chang B, Ewing CM, Bujnovszky P, Carpten JD, Bleecker ER, Walsh PC, Trent JM, Meyers DA, Isaacs WB (2001) Linkage and association studies of prostate cancer susceptibility: evidence for linkage at 8p22-23. Am J Hum Genet 69: 341–350
Xu J, Zheng SL, Komiya A, Mychaleckyj JC, Isaacs SD, Hu JJ, Sterling D, Lange EM, Hawkins GA, Turner A, Ewing CM, Faith DA, Johnson JR, Suzuki H, Bujnovszky P, Wiley KE, DeMarzo AM, Bova GS, Chang B, Hall MC, McCullough DL, Partin AW, Kassabian VS, Carpten JD, Bailey-Wilson JE, Trent JM, Ohar J, Bleecker ER, Walsh PC, Isaacs WB, Meyers DA (2002) Germline mutations and sequence variants of the macrophage scavenger receptor 1 gene are associated with prostate cancer risk. Nat Genet 32: 321–325
Acknowledgements
We thank Minna Sjöblom and Riitta Vaalavuo for excellent technical assistance. The prostate cancer patients and their families are thanked for their participation and cooperation. This study was supported by the Medical Research Foundation of Tampere University Hospital, the Reino Lahtikari Foundation the Finnish Cancer Organisations, the Sigrid Juselius Foundation and the Academy of Finland. EHS received support from Pirkanmaa Cancer Society and from the University of Tampere.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Seppälä, E., Ikonen, T., Mononen, N. et al. CHEK2 variants associate with hereditary prostate cancer. Br J Cancer 89, 1966–1970 (2003). https://doi.org/10.1038/sj.bjc.6601425
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6601425
Keywords
This article is cited by
-
Association of CHEK2 I157T and SULT1A1 R213H genetic variants with risk of sporadic colorectal cancer in a sample of Egyptian patients
Egyptian Journal of Medical Human Genetics (2022)
-
An appraisal of genetic testing for prostate cancer susceptibility
npj Precision Oncology (2022)
-
Risk of developing a second primary cancer in male breast cancer survivors: a systematic review and meta-analysis
British Journal of Cancer (2022)
-
Genetic predisposition to prostate cancer: an update
Familial Cancer (2022)
-
Cell-type-specific role of CHK2 in mediating DNA damage-induced G2 cell cycle arrest
Oncogenesis (2020)
