
Abstract
Duchenne and Becker muscular dystrophies (DMD/BMD) are the most commonly inherited neuromuscular disease. However, accurate and convenient molecular diagnosis cannot be achieved easily because of the enormous size of the dystrophin gene and complex causative mutation spectrum. Such traditional methods as multiplex ligation-dependent probe amplification plus Sanger sequencing require multiple steps to fulfill the diagnosis of DMD/BMD. Here, we introduce a new single-step method for the genetic analysis of DMD patients and female carriers in real clinical settings and demonstrate the validation of its accuracy. A total of 89 patients, 18 female carriers and 245 non-DMD patients were evaluated using our targeted NGS approaches. Compared with traditional methods, our new method yielded 99.99% specificity and 98.96% sensitivity for copy number variations detection and 100% accuracy for the identification of single-nucleotide variation mutations. Additionally, this method is able to detect partial deletions/duplications, thus offering precise personal DMD gene information for gene therapy. We detected novel partial deletions of exons in nine samples for which the breakpoints were located within exonic regions. The results proved that our new method is suitable for routine clinical practice, with shorter turnaround time, higher accuracy, and better insight into comprehensive genetic information (detailed breakpoints) for ensuing gene therapy.
Similar content being viewed by others

Introduction
Duchenne muscular dystrophy (DMD, MIM no. 310200) is one of the most common progressive and severely disabling neuromuscular diseases of childhood. It is an X-linked recessive disorder affecting ∼1 in 3500 live male newborns.1 DMD and its milder allelic Becker muscular dystrophy (BMD, MIM no. 300376) are caused by mutations in the dystrophin (DMD) gene.2 The DMD gene, one of the largest genes identified to date, contains 79 exons, 78 introns, and 8 promoters, spanning more than 2.5 Mb of genomic DNA. DMD/BMD are caused by a number of different types of mutations, including deletions (∼60%) or duplications (∼7%) of one or more exons, small insertions or deletions within an exon (∼7%), single-nucleotide point mutations (∼20%), and splice site or intronic mutations (<1%).3 Most laboratories perform two or more steps to detect mutations in a suspected DMD patient. Multiplex ligation-dependent probe amplification (MLPA) or array comparative genome hybridization (aCGH) is used to detect large deletions/duplications;4, 5 if no deletion/duplication is detected, Sanger sequencing of all the DMD exons is performed.6, 7 However, in addition to the high cost and time requirements of the entire process, the results can be inconclusive.
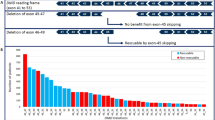
Recently, several promising strategies that target the primary genetic defect of dystrophin have entered clinical trials, and the most advanced therapy targets only specific mutation types.8, 9, 10, 11, 12 Exon skipping, which rectifies the aberrant reading frame, aims to treat patients with large deletions; yet, to achieve this, the selection of appropriate patients with detailed genetic diagnosis is very important.
In the past few years, targeted NGS approaches have become an important tool in identifying disease-causing genes and making clinical diagnoses based on the bioinformatic analysis of massive parallel DNA-sequencing efforts.13, 14 Lim et al.15 reported a mutational search platform for the genetic diagnosis of DMD/BMD, but only a small number of samples was included; these authors only reported the detection of large deletion/duplication mutations in male patients. In the present study, we developed a novel computational framework and applied this platform to identify copy number variations (CNVs) and single-nucleotide variations (SNVs) in a large cohort at the same time. Additionally, this method is able to provide detailed locations of the breakpoints. For CNVs, the results obtained from targeted NGS approaches were compared with MLPA; for SNVs and insertion/deletions, the results were validated by Sanger sequencing. The targeted NGS approaches offered more detailed information of the mutated dystrophin gene, enabling us to understand the molecular pathogenic mechanism better and to identify more suitable candidate patients for emerging gene therapy.
Materials and methods
Study participants
The study was divided into two phases (Supplementary Figure S1). Phase I aimed to establish the targeted NGS approaches for the genetic diagnosis of DMD/BMD, and Phase II aimed to validate the efficacy of the targeted NGS approaches in real clinical practice. Written informed consent was obtained from all the participants when collecting their peripheral blood. The institutional Ethics Committee of the PUMCH approved the study protocol (PUMCH S-411).
In Phase I, 124 healthy volunteers were recruited by BGI-Shenzhen, Shenzhen (South China), and their data were used to set up the normal range and SD of certain DMD exons. Then, a group of 30 retrospective DMD/BMD patients and female carriers were enrolled, and their diagnoses were confirmed by immunohistochemical staining of dystrophin in muscle biopsies by Peking Union Medical College Hospital (PUMCH), Beijing (North China) and Zhejiang University School of Medicine Women’s Hospital (ZUSMWH), Hangzhou (South China). The blood samples of the DMD/BMD patients and carriers were sent to BGI for targeted NGS approaches and PUMCH for MLPA detection. If a disease-causing SNV was identified by the targeted NGS approaches, Sanger sequencing was conducted by BGI to validate the result. The investigators from BGI and PUMCH were blinded to the results until the final unblinding. In Phase II, 80 clinically diagnosed DMD/BMD patients, female carriers and 245 non-DMD patients were recruited by PUMCH and ZUSMWH in a consecutive manner during a 6-month period from the 1 April 2011 to 30 September 2011. The blood samples of the DMD/BMD patients, female carriers, and 245 non-DMD patients were sent to BGI for targeted NGS approaches; these blood samples were also sent to PUMCH for simultaneous MLPA detection. If a disease-causing SNV was identified, Sanger sequencing was conducted by BGI to validate the result (Supplementary Table S5). Upon the completion of this step, the sensitivity and specificity of the CNV detection by the targeted NGS approaches were calculated in the data center for statistics. In Phase II, two DMD patients were excluded because they were later diagnosed as having other neuromuscular disorders (one inflammatory myopathy and one limber-girdle muscular dystrophy). Two female DMD patients, with clinical manifestations, lab test, and muscle biopsies that were consistent with typical DMD patients, were also recruited in this phase.
Workflow of targeted NGS approaches
First, the peripheral blood samples of the participants were analyzed using targeted NGS approaches, with 10–33 samples being analyzed together in a pooled batch. The bioinformatics process began after the generation of raw data by Illumina Pipeline (version 1.3.4). Bam data, the output file after alignment to the reference human genome, were used for the following analysis. The reference value from the data of 124 healthy volunteers was then established. CNVs were detected using a three-step computational framework for the 89 DMD/BMD patients, 18 female carriers and 245 non-DMD patients. Lastly, SNV and INDEL detection was performed for the DMD/BMD patients, female carriers and non-DMD patients (Figure 1).

Workflow of the targeted NGS approaches. (a) Establishment of the reference value using the data from 124 healthy volunteers. (b) CNV detection using a three-step computational framework for the DMD/BMD patients, female carriers, and non-DMD samples. (c) Formula used in the targeted NGS approaches. *ND_exonN/ND_base: normalized sequencing depth of certain exons or single bases in the DMD gene of a certain sample; †ExonN_depth: sequencing depth of a certain exon in the DMD gene of a certain sample; ‡WTR_depth: sequencing depth of the entire targeted region of 222 genes in a certain sample; Mean ND_exonN batch's control, mean of ND_exonN of all barcoded samples from the same batch.
For description of the criteria for DMD/BMD patient recruitment, targeted next-generation sequencing approach, three-step computational framework for CNVs, MLPA procedure and Sanger sequencing, see Supplementary Texts.
Results
Reference value establishment and three-step computational framework for CNV detection
To establish the reference value, the bam data from the 124 healthy volunteers was used to calculate the normalized sequencing depth of all 79 DMD exons (ND_exonN) by the targeted NGS approaches. The mean and SD ND_exonN values of the healthy volunteers were also calculated. The mean ND_exonN for each exon in males was approximately half of that in females, which was coincident with the copy number of the DMD gene.
The intra-batch ratios of the healthy volunteers were also calculated. Specifically, the intra-batch ratio was defined as ND_exonN divided by the mean ND_exonN of all samples in a batch. The intra-batch ratios were useful to filter out false-positive exons during CNV detection.
The intra-batch ratios of the male and female healthy volunteers were subjected to Gaussian distribution and plotted (Figures 2a and b). The mean of the intra-batch ratios of the male and female volunteers is equal to 1.0000, whereas the SD were 0.106477 and 0.092178, respectively. It is postulated that the intra-batch ratio of true CNV exons should at least exceed four times the SD of the Gaussian distribution of healthy volunteers of each gender. The cutoff values were four times the SD, which were 1.43, 0.63, and 1.37 for the male duplication, female deletion, and female duplication, respectively. In detail, the true male exon duplication should be more than 1.43, the true female exon deletion should be less than 0.63, and true female exon duplication should be more than 1.37.

Distributions of the intra-batch ratio of healthy volunteers and distributions of the ND_exonN values of DMD/BMD patients, female carriers, and non-DMD samples. (a) Distribution of the intra-batch ratio of male healthy volunteers. The cutoff value should be >1.43 for the male exon duplications. (b) The distribution of the intra-batch ratio of the female healthy volunteers. The cutoff value should be >1.37 for the female exon duplications. The cutoff value should be <0.63 for the female exon deletions. (c) The ND_exonN value of all 79 exons of the DMD/BMD patients, female carriers, and non-DMD patients. Each point indicates an exon. Orange triangles: female exons. Blue triangles: male exons in which dark blue ones indicate male exon deletions.
For all 79 exons of the DMD gene, the ND_exonN values of both male and female DMD/BMD patients, carriers and non-DMD samples were plotted. Excluding male deletions, the general distribution pattern perfectly matches that of the reference group (Figure 2c, Supplementary Table S3). The data were then subjected to the three-step computational framework. The male exonic deletions were detected based on the ND_exonN value (Figure 2c), whereas the male partial deletions were detected by the ND_base value. Seven partial deletions were found: exon 45 of P06, P07, P08, P09, and P10, exon 3 of P29, and exon 44 of P67 (Figure 2c, Table 2). The MLPA results of all the male deletions and partial deletions were positive. The male duplications and female deletions/duplications were then detected. To clearly depict the distribution of CNVs versus normal exons, different colors were used to indicate the different types of CNVs and normal exons (Figures 3a–f). All the exons for the male duplications, female deletions, and female duplications were first filtered by Z_score, yielding some candidate exons. When calculating Z_score, two false-negative results occurred (C35, exon 9, with a Z_score=−0.57; C02, exon52, with a Z_score=−2.53, shown in Figure 3a). Those candidate exons were then filtered by Z_scoreR, yielding fewer candidate exons. When calculating Z_scoreR, one more false-negative results occurred (C12, exon 44, with a Z_scoreR=3.71, shown in Figure 3c). C57, exon 46, with a Z_scoreR=3.69, was verified as a partial deletion and not a true false-negative result. Lastly, those candidate exons were filtered by intra-batch ratios, yielding true CNVs. One more false-negative result (C11, exon 44, with an intra-batch ratio of 1.29, shown in Figure 3e) and two false-positive results (P45, exon18 with an intra-batch ratio of 1.47; P09, exon2 1.54, shown in Figure 3f) occurred. The three filters mentioned above should maximally reduce the false-positive results.

Distributions of the Z_score, Z_scoreR values, and intra-batch ratios of DMD/BMD patients, female carriers, and non-DMD samples. (a, b) Z_score for all 79 exons of the DMD/BMD patients, female carriers, and non-DMD samples. Red triangles, true exon duplications. Blue triangles, true exon deletions. Dots of different colors, normal exons without duplications or deletions. (c, d) Z_scoreR of candidate exons from the previous step. (e, f) Intra-batch ratios of candidate exons from the previous step.
After all three steps, 68 individuals (54 patients and 14 carriers) among 89 DMD/BMD patients and 18 female carriers were detected as having CNVs (Supplementary Figures S2 and S3, Table 2). No individuals were detected as having CNVs among the 245 non-DMD patients.
Duplication/deletion breakpoint analysis
The ten-base pair-flanking sequence on either side of all exons was also covered by our method. The normalized depth of the 5′- and 3′- flanking sequences of the 79 exons were obtained using a similar approach as previously mentioned. If a significant difference between two adjacent segments was observed, the depth-decreased region would be presumed to contain a breakpoint. So, the detailed positions of the breakpoints of each CNV exon from the previous steps were determined. For instance, P01 had a significant difference between the 3′-flanking sequence of exon 45 and 5′-flanking of exon 46. Therefore, the breakpoint should be located within intron 45. Similarly, another breakpoint was found in intron 55 (Figure 4a), and we localized the breakpoints in C01 (P01’s mother) to be within intron 45 and intron 55 (Figure 4b). Figures 4c and d show the breakpoints in P59 and C59 to be within intron 1 and intron 3.

Duplication/deletion breakpoint analysis. (a–d) Four individuals (P01, C01, P59, and C59) with breakpoints located in intronic regions. Yellow columns, ND_exonN. Blue columns, normalized depth of 5′-flanking sequences. Red columns, normalized depth of 3′-flanking sequences. (e) P06 with endpoint located within exon 45. Note the significant difference between the 3′-flanking sequences of exon 45 and 5′-flanking sequences of exon 45. (f) Detailed analysis, with the single-nucleotide sequencing depth revealing that the endpoint of P06 was near c.6520 in exon 45. (g) The endpoint of C57 was located within exon 46. Note the significant difference between the 3′-flanking sequences of exon 46 and 5′-flanking sequences of exon 46. (h) Detailed analysis, with the single-nucleotide sequencing depth revealing that the endpoint of C57 was near c.6656 in exon 46.
If the breakpoint was located in an exonic region, it was defined as a partial deletion. For male deletions, a partial deletion was already detected in the preceding step 1. For the male duplications, female deletions, and female duplications, a partial deletion was detected if a significant difference between the 3′-flanking and 5′-flanking sequences of the same exon was detected.
In total, nine exons with partial deletions were detected, including the members of a pedigree of P06 (P07-10 and C06) and other unrelated subjects (P29, P67, and C57). The details of the flanking sequences of all CNV samples are shown in Supplementary Figure S4.
Interestingly, P06, who was determined to have an exon 45 deletion by MLPA and had an ND_exonN for exon 45>0.1, showed no read depth in 5’-flanking of exon 45, partial read depth in exon 45, and a normal read depth in 3′-flanking of exon 45, indicating that the endpoint of deletion was located within exon 45 (Figure 4e). We then analyzed the single-nucleotide sequencing depth. The results indicated that the breakpoint should be at approximately c.6520 in exon 45 (Figure 4f). C57 was determined to have an exon 45_46 deletion by MLPA. A significant increment between the 5′-flanking of exon 46 and the 3′-flanking of exon 46 was detected (Figure 4g). Therefore, the endpoint was located in exon 46, and the single-nucleotide sequencing depth validated this finding (Figure 4h).
Detection of SNVs and INDELs
A total of 37 patients and carriers were found to have SNVs or INDELs, and all detected SNVs and INDELs in the DMD gene were distributed in three of four functional domains (Table 3, Supplementary Figures S5 and S6). There were 12 INDELs, 18 nonsense mutations, 3 missense mutations and 4 splice site mutations. Altogether, 24 mutations were novel. All the SNVs and INDELs were validated by Sanger sequencing.
Statistical analysis of the targeted NGS approaches
In this study, the average coverage of the samples of 476 participants (124 healthy volunteers, 89 DMD/BMD patients, 18 female carriers, and 245 non-DMD patients) was 99.70%, and the lowest coverage was 98.45%. We also computed the 20 × coverage (the coverage of sites with depth greater than 20 in the targeted region); the average 20 × coverage was 98.75%, and the lowest 20 × coverage was 95.78%. The average sequencing depth of all the samples was 352.46, with a SD of 181.28; the lowest sequencing depth was 91.71 (Supplementary Tables S1 and S2).
The CNV results from the targeted NGS approaches were compared with the results using MLPA. The overall accuracy of the CNV results was calculated as the number of completely matched individuals divided by the total number of DMD/BMD patients, carriers, and non-DMD samples. The completely matching individuals were defined as whose CNV result from both the targeted NGS approaches and MLPA were completely matched. Compared with the MLPA results, there were four false-negative and two false-positive results. Thus, the overall accuracy was 98.30% (346/352) at the participant level (Table 1).
The sensitivity and specificity of the CNV results were also calculated at the exonic level. For the 89 DMD/BMD patients, 18 female carriers, and 245 non-DMD patients, the total exon number was 27 808 (352 × 79), and 384 exons were identified by both the targeted NGS approaches and MLPA. Four false-negative and two false-positive results were found. The sensitivity was 98.96% (384/388) and specificity was 99.99% (27 418/27 420) (Tables 1, 2 and 3).
Among 89 DMD/BMD patients who underwent targeted NGS approaches, two patients (P80 and P86) could not be identified with a disease-causing CNV, SNV, or INDEL. Even though their clinical manifestations, family history, lab tests and immunohistochemical staining of muscle biopsy were consistent with DMD. We suspect that there might be a defect in a deep intron, which requires further investigation.
Discussion
In 1986, Kunkel et al.16 identified the locus of the DMD gene, and clinicians have since attached a greater importance to the molecular diagnosis of DMD/BMD. In 1988, Chamberlain et al.17 first designed an assay using multiplex PCR and electrophoresis to detect several deletion-prone exons. Currently, MLPA and aCGH are the most widely applied methods for the detection of large deletions and duplications.18 However, none of these techniques can detect SNVs. Thus, if no deletions or duplications are found, the samples must be subjected to a second test of Sanger sequencing, increasing the cost and turnaround time.
The high demand for low-cost and high-throughput sequencing has led to the development of NGS.19 Recent studies applying NGS have proven its ability in detecting SNVs and large deletions/duplications in the dystrophin gene in males.15, 20 However, the detection of CNV in female carriers by targeted NGS approaches had not been reported to date. Compared with haploid X chromosomes in male, the CNV detection in females with diploid X chromosomes would be more complex. Accordingly, we attempted to design an approach to detect CNV in both patients with and female carriers of this disease.
The novel three-step computational framework is able to detect exon-level CNVs. First, the normal ranges of the normalized sequencing depth for all exons based on the reference group were established. The sequencing depth of a mutated exon would be out of the range of the Gaussian distribution, but lie within the range after rectification by the theoretical CFs.21 However, due to inevitable inter-batch differences and variability of targeted NGS approaches, some false-positive results were intermingled with true CNVs. Therefore, we used the intra-batch ratio to further distinguish the CNVs from false-positive results (step 3). The normal reference intra-batch ratio fitted the Gaussian distribution very well. We postulated that an exon with a ratio exceeding four times the SD of the Gaussian distribution of healthy volunteers of the same gender to be a true CNV. There were still four false-negative results, which were all from female carriers, and most of them were single mutated exons or marginal exons. Although the results indicate that targeted NGS approaches could detect CNVs in female carriers, this detection is more difficult, and further refinement and improvement of the assay is required for testing in female carriers and other targeted genes located in autosomal chromosomes.
In addition to the large amount of participants, our study verified the practical efficacy of targeted NGS approaches in genetic diagnoses of consecutive clinically suspected DMD/BMD patients and female carriers without any selection. Thereafter, our platform was applied in routine tests. Thus, it is a successful example of translating new technologies into clinical practice.
We first systemically report a new subtype of CNV with breakpoints located in exons. Limited by the inability of previous methods to provide detailed information about the breakpoints, clinical geneticists have postulated that breakpoints are always located in introns. However, based on our study, breakpoint in exons is not an event with a small probability. Even when regarding a pedigree as 1 person, there were 4 persons with partial deletions in 52 deletion patients and female carriers. The difference between partial and total exon deletion clearly influences the phenotype of patients and the therapeutic outcome of exon-skipping therapy. In our study, a 59-year-old female carrier was diagnosed as having an exon 45_46 deletion by MLPA and total deletion of exon 45 and partial deletion of exon 46 by targeted NGS approaches. This woman had two sons manifested as typical DMD, and both of them experienced onset at 3 years of age, progressed to non-ambulant stage by 11 and died at 18. According to the reading-frame principle, the deletion of exons 45 and 46 is apparently an in-frame mutation, and the patients should have a milder phenotype. Actually, the mutation of this pedigree is out-of-frame due to the partial deletion of exon 46. Approximately 7–9% of patients with dystrophin gene deletions disagree with the reading-frame rule.8 We suggest that this type of inaccurate judgment accounts for at least a part of the discordance between genotype and phenotype.
In recent years, several promising novel gene-reframing strategies that require detailed information about the mutated gene have entered clinical trials.8 Gentamycin and ataluren (PTC24), allowing nonsense mutation reversion, have completed phase II clinical trials.9, 10 The antisense oligonucleotides, such as AVI-4658 and PRO051, which induce exon 51 skipping, have completed phase II clinical trials; PRO051 is currently undergoing a phase III clinical trial.11, 12 Because all of these molecules target specific mutation types, the detailed location of a breakpoint is important in selecting appropriate participants for exon-skipping treatment. Obviously, partial exonic deletion may alter the therapeutic effect of these therapies. For example, the pedigree of P06, who was diagnosed by MLPA as having an exon 45 deletion, apparently could be treated with exon 44 skipping. However, the breakpoint analysis showed the presence of the remaining exon 45 sequence and the splicing region, which would influence the therapeutic effect of exon 44 skipping. We even verified the intra-exonic breakpoint and determined the breakpoint in P06’s pedigree to be at approximately c.6520 by the single-base ND values. Moreover, the method provides accurate SNP information within the antisense oligonucleotide-binding sites, which could possibly alter the binding affinity of antisense oligonucleotides. In the future, personalized therapeutic strategies can be designed based on precise DMD gene information.
There were two female DMD patients. Although their clinical manifestation, family history and muscle biopsy were typical for Duchenne muscular dystrophy, their genotypes were consistent with female carriers. In particular, the monozygotic twin (C36) of one female patient (P36) manifested as a carrier. We believe that the reason why these two female patients suffered from dystrophinopathy is not limited to DNA defects. There must be other mechanisms influencing the pathogenesis.
Carrier screening may reduce the incidence of DMD and ameliorate the suffering associated with DMD.22 DMD female carriers are prone to developing cardiomyopathy, which can be effectively prevented and treated if the carriers are diagnosed early and followed-up routinely.23 Indeed, many experts advocate DMD screening in all newborns for diagnosis as early as possible.24 Our platform can be used as a promising tool for screening asymptomatic female carriers and newborns. In the near future, with such new platforms as Miseq or Ion Torrent PGM, the testing cycle could be shortened to just a few days.25 Furthermore, the cost of analyzing an individual sample is much lower than that using MLPA plus Sanger sequencing.
The main limitation of our strategy is that deep intronic mutations and complex rearrangements may not be detected; these constitute ∼2% of the mutations in the patient population (such as P80 and P86). If the patient receives a definite diagnosis via muscle biopsy and immunopathology, and no causative mutation is found by our method, the patient should further undergo mRNA retrotranscriptional analysis.26
In conclusion, we have developed and verified a novel single test based on targeted NGS approaches to provide the comprehensive detection of mutations in DMD/BMD patients and female carriers. The extension of CNV detection from the haploid X chromosome to diploid chromosome will substantially enable this method being applied to autosome genetic diseases, such as tuberous sclerosis complex and neurofibromatosis.
References
Bushby K, Finkel R, Birnkrant DJ et al: Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 2010; 9: 77–93.
Hoffman EP, Brown RH, Kunkel LM : Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 1987; 51: 919–928.
Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT : Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006; 34: 135–144.
Lalic T, Vossen RH, Coffa J et al: Deletion and duplication screening in the DMD gene using MLPA. Eur J Hum Genet 2005; 13: 1231–1234.
Hegde MR, Chin EL, Mulle JG, Okou DT, Warren ST, Zwick ME : Microarray-based mutation detection in the dystrophin gene. Hum Mutat 2008; 29: 1091–1099.
Bennett RR, den Dunnen J, O'Brien KF, Darras BT, Kunkel LM : Detection of mutations in the dystrophin gene via automated DHPLC screening and direct sequencing. BMC Genet 2001; 2: 17.
Flanigan KM, Dunn DM, von Niederhausern A et al: Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat 2009; 30: 1657–1666.
Pichavant C, Aartsma-Rus A, Clemens PR et al: Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol Ther 2011; 19: 830–840.
Malik V, Rodino-Klapac LR, Viollet L et al: Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann Neurol 2010; 67: 771–780.
Welch EM, Barton ER, Zhuo J et al: PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007; 447: 87–91.
Cirak S, Arechavala-Gomeza V, Guglieri M et al: Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 2011; 378: 595–605.
Goemans NM, Tulinius M, van den Akker JT et al: Systemic administration of PRO051 in Duchenne's muscular dystrophy. N Engl J Med 2011; 364: 1513–1522.
Ng SB, Buckingham KJ, Lee C et al: Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 2010; 42: 30–35.
Calvo SE, Compton AG, Hershman SG et al: Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med 2012; 4: 118ra110.
Lim BC, Lee S, Shin JY et al: Genetic diagnosis of Duchenne and Becker muscular dystrophy using next-generation sequencing technology: comprehensive mutational search in a single platform. J Med Genet 2011; 48: 731–736.
Kunkel LM, Hejtmancik JF, Caskey CT et al: Analysis of deletions in DNA from patients with Becker and Duchenne muscular dystrophy. Nature 1986; 322: 73–77.
Chamberlain JS, Gibbs RA, Ranier JE, Nguyen PN, Caskey CT : Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res 1988; 16: 11141–11156.
del Gaudio D, Yang Y, Boggs BA et al: Molecular diagnosis of Duchenne/Becker muscular dystrophy: enhanced detection of dystrophin gene rearrangements by oligonucleotide array-comparative genomic hybridization. Hum Mutat 2008; 29: 1100–1107.
Hall N : Advanced sequencing technologies and their wider impact in microbiology. J Exp Biol 2007; 210: 1518–1525.
Xie S, Lan Z, Qu N et al: Detection of truncated dystrophin lacking the C-terminal domain in a Chinese pedigree by next-generation sequencing. Gene 2012; 499: 139–142.
Chiu RW, Akolekar R, Zheng YW et al: Non-invasive prenatal assessment of trisomy 21 by multiplexed maternal plasma DNA sequencing: large scale validity study. BMJ 2011; 342: c7401.
Bell CJ, Dinwiddie DL, Miller NA et al: Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med 2011; 3: 65ra64.
Politano L, Nigro V, Nigro G et al: Development of cardiomyopathy in female carriers of Duchenne and Becker muscular dystrophies. JAMA 1996; 275: 1335–1338.
Mendell JR, Shilling C, Leslie ND et al: Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol 2012; 71: 304–313.
Loman NJ, Misra RV, Dallman TJ et al: Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol 2012; 30: 434–439.
Bovolenta M, Scotton C, Falzarano MS, Gualandi F, Ferlini A : Rapid, comprehensive analysis of the dystrophin transcript by a custom micro-fluidic exome array. Hum Mutat 2012; 33: 572–581.
Acknowledgements
We thank all the blood donors for their invaluable contribution to this study. The study received financial support from Shenzhen Development and Reform Commission project, Guangdong Provincial Science and Technology Department Planning Project, Shenzhen Birth Defect Screening Project Lab (JZF No. (2011) 861) and Peking Union Medical College Hospital Young Scholars Project. Both of the projects are non-profit research projects by government and institutions.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Wei, X., Dai, Y., Yu, P. et al. Targeted next-generation sequencing as a comprehensive test for patients with and female carriers of DMD/BMD: a multi-population diagnostic study. Eur J Hum Genet 22, 110–118 (2014). https://doi.org/10.1038/ejhg.2013.82
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.82
Keywords
This article is cited by
-
Identification of two novel large deletions in FBN1 gene by next-generation sequencing and multiplex ligation-dependent probe amplification
BMC Medical Genomics (2024)
-
A female patient carrying a novel DMD mutation with non-random X-chromosome inactivation from a DMD family
BMC Medical Genomics (2024)
-
DMD/BMD prenatal diagnosis and treatment expectation in a single centre in China for 15 years
BMC Medical Genomics (2021)
-
Complexity of skeletal muscle degeneration: multi-systems pathophysiology and organ crosstalk in dystrophinopathy
Pflügers Archiv - European Journal of Physiology (2021)
-
Mutation Spectrum of Dystrophinopathies in India: Implications for Therapy
The Indian Journal of Pediatrics (2020)
