
Abstract
We have identified a deletion of 3 base pairs in the dystrophin gene (DMD), c.9711_9713del, in a family with nonspecific X-linked intellectual disability (ID) by sequencing of the exons of 86 known X-linked ID genes. This in-frame deletion results in the deletion of a single-amino-acid residue, Leu3238, in the brain-specific isoform Dp71 of dystrophin. Linkage analysis supported causality as the mutation was present in the 7.6 cM linkage interval on Xp22.11–Xp21.1 with a maximum positive LOD score of 2.41 (MRX85 locus). Molecular modeling predicts that the p.(Leu3238del) deletion results in the destabilization of the C-terminal domain of dystrophin and hence reduces the ability to interact with β-dystroglycan. Correspondingly, Dp71 protein levels in lymphoblastoid cells from the index patient are 6.7-fold lower than those in control cell lines (P=0.08). Subsequent determination of the creatine kinase levels in blood of the index patient showed a mild but significant elevation in serum creatine kinase, which is in line with impaired dystrophin function. In conclusion, we have identified the first DMD mutation in Dp71 that results in ID without muscular dystrophy.
Similar content being viewed by others


Introduction
The dystrophin gene (DMD) is the largest gene in the human genome described to date, spanning >2.2 Mb (UCSC genome Browser, Hg19). Its largest isoform, Dp427m, consists of 79 exons and 3685 amino-acid residues.1 DMD in total comprises 86 exons, as for the different isoforms there are eight different first exons possible. The main isoforms are Dp427, Dp260, Dp140, Dp116 and Dp71 named after their protein molecular weight. The expression of these isoforms is driven by specific promoters for brain, muscle and retina. Accordingly, the expression of the isoforms is directed to specific tissues: Dp427 can be expressed in skeletal and cardiac muscle (Dp427m), brain (Dp427b) and Purkinje cells (Dp427p). Dp260 is almost exclusively expressed in retina,2 whereas the Dp140 is expressed in brain, retina and kidney3 and Dp116 only in adult peripheral nerves.4 Remarkably, the shortest isoform, Dp71, is expressed in most tissues, such as brain, retina, kidney, liver, lung and heart, but not in muscle.5
DMD encodes a cytoskeletal protein, dystrophin, that links intracellular γ-actin to the dystrophin-associated glycoprotein complex in the cell membrane in both muscle and nonmuscle tissues.6 In muscle, dystrophin is essential for maintaining the structural integrity of the sarcolemma. Consequently, the absence of functional dystrophin results in increased membrane permeability that can be measured by the release of soluble muscle enzymes, such as creatine kinase, in the blood. In neurons, the dystrophin-associated glycoprotein complex is involved in postsynaptic clustering of ion channels and postsynaptic membrane receptors during synaptogenesis.7 The presence of dystrophin and the dystrophin-associated glycoprotein complex in synapses in distinct brain areas, such as the cerebral cortex, hippocampus and cerebellum,8, 9 suggests that loss-of-function may be responsible for the cognitive deficits associated with mutations in DMD.
Mutations almost invariably result in either Duchenne muscular dystrophy or the milder Becker muscular dystrophy. The muscular dystrophy is often accompanied by intellectual disability (ID), especially in case of truncating mutations that lead to the absence of Dp71.10, 11 To our knowledge, only one patient has been described with a DMD mutation with ID, but without muscular dystrophy: a deletion of exons 3–9 affecting only the largest dystrophin isoform Dp427.12 Here, we describe a family of six males who have nonspecific X-linked ID (XLID; MRX85), with a DMD mutation that only disturbs the shortest brain-specific isoform, Dp71.
Patients and methods
Patients
Family N051 contains six males with non-progressive mild-to-moderate ID according to the DSM-IV criteria in two generations (Figure 1). Karyotypes at a resolution of 550 bands were normal for these family members and expansions of the CGG repeat in the 5′-untranslated region of FMR1, which cause the Fragile X syndrome, were excluded as well. In family member III-9, deletions or duplications of one or more exons on the X chromosome were excluded by using the chromosome X exon-specific array.13 Family member III-11 was first documented as unaffected as he attended and finished normal elementary school although he repeated two grades, could live on his own and was a gardener. Unexpectedly, upon examination at the age of 31 years, moderate ID according to the Wechsler Intelligence Scale for Children-Revised (WISC-R) and Raven's Colored Progressive Matrices (CPM) was established. His adaptive functioning was at the borderline level as measured by the ‘Sociale Redzaamheidsschaal’, a Dutch-specific test to assess adaptive functioning.14 One family member, II-5, was classified at the age of 70 years as having mild ID according to the WISC-R and CPM intelligence tests, but he attended regular elementary school, had a normal working life in industry and lived on his own. Since he was 70 years old at the time of testing, it is likely that other non-genetic factors might have influenced his IQ test scores. Pregnancy and delivery was uneventful for all patients. Besides the ID, no further anomalies were reported, although patient III-4 showed aggressive behavior. All affected males could walk and ride a bike without need for assistance. Performance during other physical exercises, such as walking stairs and throwing a ball was normal. These abilities did not regress during their lives. All obligate female carriers have normal cognitive capabilities. In summary, these examinations indicate that the patients in this family have nonspecific XLID.

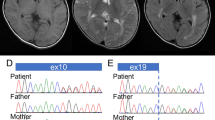
Pedigree, haplotypes and mutation analysis of family N051. (a) Pedigree and haplotypes for the linkage interval on Xp21.3-p21.1. Question marks indicate uncertainty about the affected status. The at-risk haplotype is indicated by the black bar. Brackets indicate inferred haplotypes. The marker order is according to the UCSC Genome Browser hg19 assembly. Asterisks indicate all family members who have been screened for the presence of the p.(Leu3238del) (annotation according to dystrophin isoform Dp427m; NM_004006.2) by conventional Sanger sequencing. All family members with the at-risk haplotype carried the mutations, whereas the ones with other haplotypes did not. (b) Electropherograms of affected family member III-9 showing the c.9711_9713del mutation (upper profile) and of unaffected family member III-8 (lower profile). Mutated base pairs and corresponding amino-acid residues are printed in bold.
Genotyping
DNA from lymphocytes was isolated as described by Miller et al15 Short tandem repeat markers on the X chromosome were amplified for 30 cycles at 58 °C annealing temperature and 3.0 mM MgCl2 by Invitrogen Taq polymerase according to the manufacturer’s protocol (Invitrogen, Breda, The Netherlands) using marker-specific primers that contain a M13 tail added to their 5′-ends. In the second PCR, the M13 tails are used to amplify the PCR product for 15 cycles at 54 °C and 3.0 mM MgCl2 by Invitrogen Taq polymerase using a M13 forward primer labeled with either FAM, VIC, NED or ROX and a M13 reverse primer with a GTTTCTT on the 5′-end that reduces tailing.16 Fluorescently labeled PCR products were analyzed by the ABI PRISM 3730 DNA analyzer (Applied Biosystems, Foster City, CA, USA) and the data analyzed by Genemapper (Applied Biosystems).
Linkage analysis
Two-point LOD scores were calculated with the SUPERLINK program,17 using easyLINKAGE18 as interface. Inheritance model was set at recessive and the frequency of the disease allele at 0.001. Full penetrance was assumed.
Mutation analysis
Primer sequences of all exons of ARX (GenBank ID NM_139058.2), IL1RAPL1 (GenBank ID NM_014271.3) and exon 67 of DMD (GenBank ID NM_004006.2) were designed by using the Primer3 program (Supplementary Table 1).19 PCR conditions are available on request. PCR products were sequenced using the ABI PRISM BigDye Terminator Cycle Sequencing V2.0 Ready Reaction Kit and analyzed with the ABI PRISM 3730 DNA analyzer (Applied Biosystems).
Amplification refractory mutation system
ARMS primers to amplify specifically either the wild-type or mutant allele were designed by using the Primer3 program (Supplementary Table 1).19 The wild-type or mutant alleles were amplified from 50 ng DNA by 10 U Taq polymerase (Invitrogen) in Taq buffer, 2.0 mM MgCl2, 0.25 mM dNTPs (Invitrogen), and 100 nM forward and reverse primer in a total volume of 25 μl. PCR cycling conditions consisted of (1) denaturation at 95 °C for 10 min, (2) 40 cycles of amplification by denaturation at 95 °C for 15 s, annealing at 58 °C for 15 s and elongation at 72 °C for 15 s, followed by (3) final elongation at 72 °C for 10 min. PCR products were analyzed on a 1.5% agarose gel.
Molecular modeling
The effect of the Leu3238 deletion on the structure of Dp71 was predicted by using the crystal structure of human dystrophin from Huang et al20 (PDB entry 1EG3; http://www.pdb.org/). The changed amino-acid side chains in the model were positioned using a backbone-dependent rotamer library as implemented in the YASARA program. The models were subsequently refined using the Yamber2 force field, which was previously shown to increase model accuracy.21 Coordinate files are available from the authors on request.
Cell culturing
Human B-lymphocytes were immortalized by transformation with Epstein-Barr virus (EBV) according to established procedures.22 EBV-transformed lymphoblastoid cell lines (EBV-LCLs) from patients and controls were grown at 37 °C and 7.5% CO2 in RPMI 1640 medium (Gibco, Breda, The Netherlands) containing 10% (v/v) fetal calf serum (Sigma, Zwijndrecht, The Netherlands), 1% 10 U/μl penicillin and 10 μg/μl streptomycin (Gibco), and 1% GlutaMAX (Gibco). Twenty-four hours before emetin treatment, cells are centrifuged at 200 × g for 5 min at room temperature and resuspended in fresh medium to a density of 0.7 million cells per ml. Cells were treated with 100 μg/ml emetin for 10 h. Subsequently, 5–10 million cells were harvested by centrifugation at 200 × g for 5 min at room temperature, washed with PBS and pelleted by centrifugation at 200 × g for 5 min at room temperature. Pellets for RNA isolation and western blotting were snap-frozen in liquid nitrogen.
Protein analysis
Pellets were resuspended in 200 μl lysis buffer containing 75 mM Tris-HCl pH 6.8 and 15% (w/v) sodium dodecyl sulfate and homogenized through a 21G needle. Protein concentrations were determined by the Pierce bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Samples containing 75, 38 or 19 μg of total protein were prepared in lysis buffer supplemented with 20% (v/v) glycerol, 5% (w/v) β-mercaptoethanol and 0.001% (w/v) Bromophenol blue, heated for 5 min at 95 °C and separated on 1.0 mm thick native PAGE Tris-acetate polyacrylamide gels, with a linear resolving gel gradient of 3–8% (Bio-Rad, Veenendaal, The Netherlands) by electrophoresis for 1.5 h at 75 V and 2 h at 100 V in Running Buffer (XT Tricine; Bio-Rad). PageRuler Prestained Protein Ladder (Thermo Fisher Scientific) was used as a marker. Gels were blotted onto Ready to use Trans-Blot Turbo transfer packs with the Trans-Blot Turbo transfer system (Bio-Rad) at 1.0 A and ∼25 V for 30 min. Membranes were blocked with Tris-buffered saline (TBS) containing 5% of non-fat dried milk (Elk; Campina Melkunie, Eindhoven, The Netherlands), washed in TBS plus 0.05% (v/v) Tween20 (TBS-T) and incubated overnight with a first antibody. For Dp71 monoclonal NCL-DYS2 antibody (Novocastra Laboratories Ltd, Newcastle Upon Tyne, UK) 1:100 in TBS at room temperature was used. For β-actin, mouse monoclonal (AC-15) to β-actin (ab6276; Abcam, Cambridge, UK) 1:5000 in TBS-5% milk at 4 °C was used. Membranes were washed in TBS-T and incubated with the fluorescent secondary antibody IRDye 800CW goat-α-mouse IgG (Li-Cor, Lincoln, NE, USA) in 1:5000 TBS/TBS-5% milk for 1 h at room temperature. Membranes were washed in TBS-T and TBS and protein were visualized with the Odyssey system (Li-Cor). Dp71 protein bands were quantified using the Image J software (http://rsbweb.nih.gov/) and normalized by using β-actin. There was a linear relationship between the detected amounts of both Dp71 and β-actin between the samples that contained 38 and 19 μg of total protein, of which the latter samples were used to determine differential Dp71 protein expression. The P-value was derived from the standard score (Z-value) calculated for the expression of Dp71 in EBV-LCLs from the patient as compared with the normal distribution of the expression of Dp71 in EBV-LCLs from three controls. We performed a one-sided test to reject the null hypothesis, that is, no statistically significant difference in the expression of Dp71 between patient and controls as we expected enhanced degradation of the destabilization protein. We used an alpha level of 0.05, because only one gene was assessed.
mRNA analysis
RNA was isolated from EBV-LCLs of patients and control individuals by using the NucleoSpin RNA II kit (Macherey-Nagel, Düren, Germany) according to the manufacturer’s protocols. The integrity of the RNA was assessed on 1.2% agarose gel, and the concentration and purity determined by optical densitometry. The OD260/OD230 and OD260/OD280 ratios were in between 1.8 and 2.0. Half a microgram of total RNA was transcribed into cDNA by using the iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA, USA) according to the manufacturer’s protocol. cDNA was purified by using the NucleoSpin extract II kit (Macherey-Nagel) according to the manufacturer’s protocol. SYBR Green-based real-time quantitative PCR (QPCR) expression analysis was performed on a 7500 Fast Real-Time PCR System (Applied Biosystems) by using Power SYBR Green PCR Master Mix (Applied Biosystems) according to the manufacturer’s instructions. Primers were designed by using the Primer3 program (Supplementary Table 1).19 PCR products encompassed at least one boundary between two exons. GUSB was used as reference gene.23 QPCR quantifications were performed in duplicate on the equivalent of 7.8 ng of total RNA from the first-strand synthesis, and included a water control. Experimental threshold cycles (Ct) values were within the range of cDNA dilutions used to validate the primers. The melt curves of all PCR products showed a single PCR product. All water controls were negative. Differences in expression of a gene of interest between the individual samples of the patients and five controls were calculated by the comparative Ct or 2ΔΔCt method.24, 25 The two-sided P-value was derived from the standard score (Z-value) calculated for each individual as compared with the normal distribution of the four controls. Primer sequences for reverse transcriptase PCR of Dp71 were designed by using the Primer3 program (Supplementary Table 1).19 PCR conditions are available on request. All reverse transcriptase PCR products were assessed by agarose gel analysis and subsequently sequenced using the ABI PRISM BigDye Terminator Cycle Sequencing V2.0 Ready Reaction Kit and analyzed with the ABI PRISM 3730 DNA analyzer (Applied Biosystems).
Results
Linkage analysis of family N051 with nonspecific ID was initially performed with a panel of 18 fluorescent markers distributed equally over the X chromosome. We assumed that family member III-11 was affected and that the affected status of II-5 was unknown. A maximum positive LOD score of 2.41 at θ=0.05 was calculated for marker DXS1202 (Table 1), which is above the significance threshold of 2.00 for X-linked families. Delineation of the linkage interval by additional markers revealed a 7.6 cM region on Xp22.11–Xp21.1 that is delimited on the telomeric site by DXS7110 and on the centromeric site by DXS1049 (Figure 1). All patients had the at-risk haplotype, except family member III-11, which suggests that he represents a phenocopy. The locus was assigned MRX (mental retardation, X-linked) number MRX85 by the HUGO Gene Nomenclature Committee (www.genenames.org/).
The MRX85 interval contains 38 known genes (UCSC Human Genome Browser, Hg19) of which two are known nonspecific XLID genes, ARX26 and IL1RAPL1.27 All exons of these two genes were analyzed by direct DNA sequencing, including donor and acceptor splice sites, and branch sites. One change, c.36C>T, was found in IL1RAPL1, which leads to a synonymous amino-acid residue change p.(Try12Tyr). This change is a known polymorphism, rs148060509 (dbSNP134; minor allele frequency 0.1%), and was not present in 350 control X-chromosomes. In addition, the cytosine at this position is not conserved between species as in dog, mouse and rat, a thymine is present at the same genomic position. In addition, the donor and acceptor splice sites are unaffected according to the NetGene2 and BDGP splice site prediction programs.28, 29 Thus, the c.36C>T transition is most likely not pathogenic.
Subsequent sequencing of all exons of 86 XLID genes on the X chromosome (coverage 95%) by droplet-based multiplex PCR and massive parallel sequencing,30 revealed an in-frame deletion of 3 base pairs (c.9711_9713del; p.(Leu3238del); Dp427m; NM_004006.2) in DMD (Figure 1). No other unique variants were found in the other exons of the X chromosome. The deletion was not present in 264 alleles from ethnically matched controls as shown by amplification refractory mutation system, in our in-house database containing all variants derived from 450 Dutch exomes, in the NHLBI exome sequencing project that currently consists of 5544 Caucasian exomes (average exon coverage >44; http://evs.gs.washington.edu/EVS/), and in the Leiden Open Variation Database (February 2013).31 The p.(Leu3238del) mutation is positioned in the middle of the MRX85 locus. As expected, all certainly affected family members carried the mutation. Leu3238 is present in all dystrophin isoforms, including the brain-specific Dp427c and Dp71 (Figure 2a) and is conserved down to Caenorhabditis elegans. Subsequent determination of the creatine kinase levels in blood of family member III-9 showed a mild elevation in serum creatine kinase (279 U/l; normal 30–200 U/l) indicative of mild muscle wasting.

Effect of the p.(Leu3238del) deletion on Dp71. (a) Overview of dystrophin with the isoforms and the position of the amino-acid change indicated. (b) Overall model of the Dp71 protein. The first EF domain is shown in orange and the second in blue. The protein region most affected by the deletion of Leu3238 is indicated in purple. (c) Detailed view of normal structure of the α-helix containing Leu3238. (d) Detailed view of the helix after introduction of the Leu3238 deletion. Figures were prepared by using the YASARA program (www.yasara.org).20 CH, calponin homology domains; EF, EF hand-like domain; SPEC, spectrin repeats; WW, domain with two conserved Trp residues.
The effect of the Leu3238 deletion on the structure of Dp71 was assessed by molecular modeling using the crystal structure of a human dystrophin fragment.20 Leu3238 is located in the second α-helix of the second EF domain (Figures 2b and c). The deletion of this amino-acid residue is predicted to result in sub-optimal side chain packing (Figure 2d). As this α-helix is involved in many packing interactions between the two EF domains, this most likely results in an destabilization of the C-terminal region of dystrophin that contains the two EF hand-like domains and the WW domain that interact with β-dystroglycan. Of note, this effect will probably attenuated by the low sequence complexity of the region surrounding Leu3238. The amino-acid residue sequence of the helix preceding Leu3238 is3222DQRRLGLLL3230. Owing to the presence of two arginine and four leucine side chains, the packing of the amino-acid side chain in the helix with the Leu3238 deletion remains partly similar to the wild-type structure.
The predicted destabilization of the C-terminal region of dystrophin by computer-assisted molecular modeling might result in a faster protein degradation. Therefore, we performed western blot analysis in EBV-LCLs from patient III-9 and three controls (Figure 3). This showed a nonsignificant 6.7-fold (P-value=0.08) reduction in Dp71 protein expression in the patient as compared normal. In addition, we performed QPCR analysis of Dp71 mRNA levels in EBV-LCLs from patient III-9, which indicated that the mRNA expression levels were within the normal range of expression (data not shown; P-value=0.61). Reverse transcriptase PCR confirmed the presence of the c.9711_9713del mutation in Dp71 and showed normal mRNA splicing of this DMD transcript (data not shown).

Immunodetection of DMD isoform Dp71 in total cell lysate from EBV-LCLs of three controls and patient III-9. Dp71 protein levels were normalized against β-actin. Dp71 expression levels in the patient are 15% of the average expression levels of the three controls.
Discussion
We have identified a mutation in DMD, p.(Leu3238del), in a family with X-linked nonspecific ID. It was the only unique variation found in all sequenced exons of the X chromosome and localized within the linkage interval in this family. Leu3238 is fully conserved down to Caenorhabditis elegans and an important amino-acid residue in the second of the two EF hand-like sequences (Pfam: PF09069) that are involved in stabilization of the WW domain.20 The EF hand-like domains and the WW domain are both part of the ∼260 amino-acid residues cysteine-rich C-terminal region that interacts with a proline-rich motif in the C-terminal tail of β-dystroglycan,32, 33 connecting dystrophin to the dystrophin-associated glycoprotein complex in the cell membrane. Molecular modeling showed that it is likely that our mutation results in the destabilization of the C-terminal domain of dystrophin and hence reduces the ability to interact with β-dystroglycan resulting in the elevated levels of serum creatine kinase measured in the patients. In line with the predicted destabilizing effect of the Leu3238 deletion on dystrophin, Dp71 protein levels in a EBV-LCL from the index patient are 6.7-fold lower than those in control cell lines.
The affected family members are characterized by nonspecific mild-to-moderate ID. There are no indications of a muscular phenotype even at an advanced age (the oldest patient is now 63 years old). Considering that our mutation affects all isoforms including the two isoforms that are proven to be involved in the reduced cognitive abilities of a number of patients with DMD mutations,10, 11 we think that our mutation only mildly impairs the function of dystrophin, thereby having only a subclinical muscular phenotype, but still resulting in a relatively large effect on the cognitive abilities of the affected family members. Previously, one other single-amino-acid deletion has been reported, p.(Glu3367del), that is found C-terminally of our mutation and supposedly initiates rotational and/or translational displacement a-helix region adjacent to the ZZ domain of DMD.34 Although plasma CK levels were high (14 724 U/l at the age of 18 months), his motor ability score at the age of 6 years old was well above average (38/40) for a boy with Duchenne muscular dystrophy of that age. These and our results indicate that single-amino-acid deletions within Dp71 result in partially functional dystrophin and hence a relatively mild phenotype ranging from simplex ID to ID accompanied by a mild muscle phenotype.
Taken together, the conserved position of Leu3238 in the EF hand-like domain of the brain-specific dystrophin isoforms Dp427 and Dp71, the reduced Dp71 protein levels in EBV-LCLs, and the elevated creatine kinase levels in serum, indicate that p.(Leu3238del) is pathogenic. According to our knowledge, this would mean that this is the first pathogenic mutation in DMD, which affects only a single amino acid, which results in ID without muscular dystrophy.
References
Muntoni F, Torelli S, Ferlini A : Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol 2003; 2: 731–740.
Pillers DA, Bulman DE, Weleber RG et al: Dystrophin expression in the human retina is required for normal function as defined by electroretinography. Nat Genet 1993; 4: 82–86.
Lidov HG, Selig S, Kunkel LM : Dp140: a novel 140 kDa CNS transcript from the dystrophin locus. Hum Mol Genet 1995; 4: 329–335.
Byers TJ, Lidov HG, Kunkel LM : An alternative dystrophin transcript specific to peripheral nerve. Nat Genet 1993; 4: 77–81.
Bar S, Barnea E, Levy Z, Neuman S, Yaffe D, Nudel U : A novel product of the Duchenne muscular dystrophy gene which greatly differs from the known isoforms in its structure and tissue distribution. Biochem J 1990; 272: 557–560.
Allen DG, Whitehead NP : Duchenne muscular dystrophy—what causes the increased membrane permeability in skeletal muscle? Int J Biochem Cell Biol 2011; 43: 290–294.
Perronnet C, Vaillend C : Dystrophins, utrophins, and associated scaffolding complexes: role in mammalian brain and implications for therapeutic strategies. J Biomed Biotechnol 2010; 2010: 849426.
Jancsik V, Hajos F : Differential distribution of dystrophin in postsynaptic densities of spine synapses. Neuroreport 1998; 9: 2249–2251.
Lidov HG, Byers TJ, Watkins SC, Kunkel LM : Localization of dystrophin to postsynaptic regions of central nervous system cortical neurons. Nature 1990; 348: 725–728.
Daoud F, Angeard N, Demerre B et al: Analysis of Dp71 contribution in the severity of mental retardation through comparison of Duchenne and Becker patients differing by mutation consequences on Dp71 expression. Hum Mol Genet 2009; 18: 3779–3794.
Taylor PJ, Betts GA, Maroulis S et al: Dystrophin gene mutation location and the risk of cognitive impairment in Duchenne muscular dystrophy. PLoS One 2010; 5: e8803.
Srour M, Bejjani BA, Rorem EA, Hall N, Shaffer LG, Shevell MI : An instructive case of an 8-year-old boy with intellectual disability. Semin Pediatr Neurol 2008; 15: 154–155.
Bashiardes S, Kousoulidou L, van Bokhoven H et al: A new chromosome x exon-specific microarray platform for screening of patients with X-linked disorders. J Mol Diagn 2009; 11: 562–568.
Kraijer DW, Kema GN : SRZ-P Sociale Redzaamheidsschaal Verstandelijk gehandicapten Hoger Niveau. Amsterdam, The Netherlands: Pearson Assessment and Information B.V., 2004.
Miller SA, Dykes DD, Polesky HF : A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
Brownstein MJ, Carpten JD, Smith JR : Modulation of non-templated nucleotide addition by Taq DNA polymerase: primer modifications that facilitate genotyping. Biotechniques 1996; 20: 1004–1010.
Fishelson M, Geiger D : Exact genetic linkage computations for general pedigrees. Bioinformatics 2002; 18 (Suppl 1): S189–S198.
Lindner TH, Hoffmann K : EasyLINKAGE: a PERL script for easy and automated two-/multi-point linkage analyses. Bioinformatics 2005; 21: 405–407.
Rozen S, Skaletsky H : Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 2000; 132: 365–386.
Huang X, Poy F, Zhang R, Joachimiak A, Sudol M, Eck MJ : Structure of a WW domain containing fragment of dystrophin in complex with beta-dystroglycan. Nat Struct Biol 2000; 7: 634–638.
Krieger E, Koraimann G, Vriend G : Increasing the precision of comparative models with YASARA NOVA—a self-parameterizing force field. Proteins 2002; 47: 393–402.
Wall FE, Henkel RD, Stern MP et al: An efficient method for routine Epstein-Barr virus immortalization of human B lymphocytes. In Vitro Cell Dev Biol Anim 1995; 31: 156–159.
de Brouwer AP, van Bokhoven H, Kremer H : Comparison of 12 reference genes for normalization of gene expression levels in Epstein-Barr virus-transformed lymphoblastoid cell lines and fibroblasts. Mol Diagn Ther 2006; 10: 197–204.
Livak KJ, Schmittgen TD : Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001; 25: 402–408.
Pfaffl MW : A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001; 29: e45.
Bienvenu T, Poirier K, Friocourt G et al: ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Hum Mol Genet 2002; 11: 981–991.
Carrie A, Jun L, Bienvenu T et al: A new member of the IL-1 receptor family highly expressed in hippocampus and involved in X-linked mental retardation. Nat Genet 1999; 23: 25–31.
Brunak S, Engelbrecht J, Knudsen S : Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol 1991; 220: 49–65.
Reese MG, Eeckman FH, Kulp D, Haussler D : Improved splice site detection in Genie. J Comput Biol 1997; 4: 311–323.
Hu H, Wrogemann K, Kalscheuer V et al: Mutation screening in 86 known X-linked mental retardation genes by droplet-based multiplex PCR and massive parallel sequencing. Hugo J 2009; 3: 41–49.
Aartsma-Rus A, van Deutekom JC, Fokkema IF, van Ommen GJ, den Dunnen JT : Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006; 34: 135–144.
Jung D, Yang B, Meyer J, Chamberlain JS, Campbell KP : Identification and characterization of the dystrophin anchoring site on beta-dystroglycan. J Biol Chem 1995; 270: 27305–27310.
Rentschler S, Linn H, Deininger K, Bedford MT, Espanel X, Sudol M : The WW domain of dystrophin requires EF-hands region to interact with beta-dystroglycan. Biol Chem 1999; 380: 431–442.
Becker K, Robb SA, Hatton Z et al: Loss of a single amino acid from dystrophin resulting in Duchenne muscular dystrophy with retention of dystrophin protein. Hum Mutat 2003; 21: 651.
Acknowledgements
We thank the members of the family for their participation in this study. This project was financially supported by the 7th European Union Framework (grant no. 241995; GENCODYS), the Dutch Brain Foundation (grant no. 2009(2)-81 to APMdB), the Dutch Organisation for Health Research and Development (ZonMw clinical fellowship grant no. 907-00-365 to TK; VENI grant no. 700.58.410 to SBN, VIDI grant no. 917.86.319 to BBAdV).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
de Brouwer, A., Nabuurs, S., Verhaart, I. et al. A 3-base pair deletion, c.9711_9713del, in DMD results in intellectual disability without muscular dystrophy. Eur J Hum Genet 22, 480–485 (2014). https://doi.org/10.1038/ejhg.2013.169
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.169
Keywords
This article is cited by
-
Overexpression of the dystrophins Dp40 and Dp40L170P modifies neurite outgrowth and the protein expression profile of PC12 cells
Scientific Reports (2022)
-
Dystrophin genetic variants and autism
Discover Mental Health (2022)
-
Computational cognitive modeling and validation of Dp140 induced alteration of working memory in Duchenne Muscular Dystrophy
Scientific Reports (2020)
-
Neurocognitive Impairment in mdx Mice
Molecular Neurobiology (2019)
-
Dp71-Dystrophin Deficiency Alters Prefrontal Cortex Excitation-Inhibition Balance and Executive Functions
Molecular Neurobiology (2019)
