
Abstract
The most appropriate time for screening for Fabry disease (FD) is school age. For this reason, we developed non-invasive methods for measuring urinary α-galactosidase A (α-gal A) protein, using enzyme-linked immunosorbent assay (ELISA), and for globotriaosylceramide (GL-3), using tandem mass spectrometry (MS/MS). We measured these two biomarkers in the urine of previously diagnosed FD hemizygotes and heterozygotes, and in controls. All the classic FD hemizygotes were clearly distinguished from controls by either method alone, and combining the two assays produced 96% sensitivity for detecting heterozygotes. To assess the utility of these methods for screening school children and adults at high risk of FD, a pilot study was conducted. To distinguish FD from 432 controls, cut-off values for α-gal A protein and GL-3 were set at the 5th and 95th centile values of the controls, respectively. Among the high-risk patients, the measurements exceeded the cut-off values for both biomarkers in male and female subjects and were strong indicators for Fabry hemizygotes and heterozygotes. However, we recommend that if the results of the first measurements exceed the cut-off values for only one of these biomarkers, another urine sample should be requested for re-assay to confirm the result.






Similar content being viewed by others
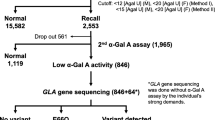

Abbreviations
- FD:
-
Fabry disease
- ERT:
-
enzyme replacement therapy
- α-gal A:
-
α-galactosidase A
- GL-3:
-
globotriaosylceramide
- MS/MS:
-
tandem mass spectrometry
- M/F ratio:
-
male-to-female ratio
- PBS:
-
phosphate-buffered saline
- Cr:
-
creatinine
References
Desnick RJ, Ioannou YA, Eng CM (2001) a-Galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Valle D, Sly WS, Childs B, Knizler KW, Vogelstein B (eds) The metabolic and molecular bases of inherited disease, 8th edn. McGraw-Hill, New York, pp 3733–3774
Schiffmann R, Murray GJ, Treco D, Daniel P, Sellos-Moura M, Myers M, Quirk JM, Zirzow GC, Browski M, Loveday K, Anderson T, Gillespie F, Oliver KL, Jeffris NO, Doo E, Liang TJ, Kreps C, Gunter K, Frei K, Crutchfield K, Selden RF, Brady RO (2000) Infusion of a-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. Proc Natl Acad Sci U S A 97:365–370
Schiffmann R, Kopp JB, Austin HA 3rd, Sabnis S, Moore DF, Weibel T, Balow JE, Brady RO (2001) Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA 285:2743–2749
Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, Caplan L, Linthorst GE, Desnick RJ (2001) Safety and efficacy of recombinant human a-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med 345:9–16
Thurberg BL, Rennke H, Colvin RB, Dikman S, Gordon RE, Collins AB, Desnick RJ, O’Callaghan M (2002) Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int 62:1933–1946
Desnick RJ, Brady R, Barranger J, Collins AJ, Germain DP, Goldman M, Grabowski G, Packman S, Wilcox WR (2003) Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management and enzyme replacement therapy. Ann Intern Med 138:338–346
Siamopoulos KC (2004) Fabry disease: Kidney involvement and enzyme replacement therapy. Kidney Int 65:744–753
Wilcox WR, Banikazemi M, Guffon N, Waldek S, Lee P, Linthorst GE, Desnick RJ, Germain DP, for the International Fabry Disease Study Group (2004) Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am J Hum Genet 75:65–74
Schoenmakere GD, Chauveau D, Grünfeld J-P (2003) Enzyme replacement therapy in Anderson-Fabry’s disease: beneficial clinical effect on vital organ function. Nephrol Dial Transplant 18:33–35
Brenner BM, Grünfeld J-P (2004) Renoprotection by enzyme replacement therapy. Curr Opin Nephrol Hypertens 13:231–241
Germain DP, Waldek S, Banikazemi M, Bushinsky DA, Charrow J, Desnick RJ, Lee P, Loew T, Vedder AC, Abichandani R, Wilcox WR, Guffon N (2007) Sustained, long-term renal stabilization after 54 months of agalasidase beta therapy in patients with Fabry disease. J Am Soc Nephrol 18:1547–1557
Desnick RJ, Brady RO (2004) Fabry disease in childhood. J Pediatr 144:s20–s26
Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C, Linhart A, Sunder-Plassman G, Ries M, Beck M (2004) Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry outcome survey. Eur J Clin Invest 34:236–242
Cho ME, Kopp JB (2004) Fabry disease in the era of enzyme replacement therapy: a renal perspective. Pediatr Nephrol 19:583–593
Warnock DG (2005) Fabry disease: diagnosis and management, with emphasis on the renal manifestations. Curr Opin Nephrol Hypertens 14:87–95
Eng CM, Germain DP, Banikazemi M, Warnock DG, Wanner C, Hopkin RJ, Bultas J, Lee P, Sims K, Brodie SE, Pastores GM, Strotmann JM, Wilcox WR (2006) Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Genet Med 8:539–548
Meikle PJ, Brooks DA, Ravenscroft EM, Yan M, Williams RE, Jaunzems AE, Chataway TK, Karageorgos LE, Davey RC, Boulter CD, Carlsson SR, Hopwood JJ (1997) Diagnosis of lysosomal storage disorders: evaluation of lysosome -associated membrane protein LAMP-1 as a diagnostic marker. Clin Chem 43:1325–1335
Chang MH, Bindloss CA, Grabowski GA, Qi X, Winchester B, Hopwood JJ, Meikel PJ (2000) Saposins A, B, C and D in plasma of patients with lysosomal storage disorders. Clin Chem 46:167–174
Mills K, Johnson A, Winchester B (2002) Synthesis of novel internal standards for the quantitative determination of plasma ceramide trihexoside in Fabry disease by tandem mass spectrometry. FEBS Lett 515:171–176
Boscaro F, Pieraccini G, La Marca G, Bartolucci G, Lucerl C, Lucerl F, Moneti G (2002) Rapid quantitation of globotriaosylceramide in human plasma and urine: a potential application for monitoring enzyme replacement therapy in Anderson-Fabry disease. Rapid Commun Mass Spectrom 16:1507–1514
Suzuki K, Owada M, Kitagawa T (2002) Study on new screening method for Fabry’s disease. J Jpn Soc Mass Screen 12(2):25
Kitagawa T, Owada M, Ishige N, Suzuki K, Ohashi T, Eto Y, Mills K, Winchester B, Keutzer J (2003) Pilot study of mass-screening for Fabry’s disease (FD) by measuring globotriaosylceramide (GL-3) in whole urine samples using tandem mass spectrometry (TMS). J Inherit Metab Dis 26 [Suppl 2]:157
Fuller M, Lovejoy M, Brooks DA, Harkin ML, Hopwood JJ, Meikel PJ (2004) Immunoquantification of a-galactosidase: evaluation for the diagnosis of Fabry disease. Clin Chem 50:1979–1985
Chamoles NA, Blanco M, Gaggioli D (2001) Fabry disease: enzymatic diagnosis in dried blood spots on filter paper. Clin Chim Acta 308:195–196
Meikle PJ, Ranieri E, Simonsen H, Rozakils T, Ramsay SL, Whitfield PD, Fuller M, Christensen E, Skovby F, Hopwood JJ (2004) Newborn screening for lysosomal storage disorders: clinical evaluation of a two tier strategy. Pediatrics 114:909–916
Meikle PJ, Grasby DJ, Dean CJ, Lang DL, Bockmann M, Whittle AM, Fietz MJ, Simonsen H, Fuller M, Brooks DA, Hopwood JJ (2006) Newborn screening for lysosomal storage disorders. Mol Genet Metab 88:307–314
Li Y, Scott CR, Chamoles NA, Ghavami A, Pinto M, Turecek F, Gelb MH (2004) Direct multiplex assay of lysosomal enzymes in dried blood spots for newborn screening. Clin Chem 50:1785–1796
Branton M, Schiffmann R, Kopp JB (2002) Natural history and treatment of renal involvement in Fabry disease. J Am Soc Nephrol 13:S139–S143
Branton MH, Schiffmann R, Sabnis SG, Murray GJ, Quirk JM, Altarescu G, Goldfarb L, Brady RO, Balow JE, Austin HA III, Kopp JB (2002) Natural history of Fabry renal disease. Influence of a-galactosidase A activity and genetic mutations on clinical course. Medicine 81:122–138
Owada M, Kitagawa T (2001) Lysosomal storage diseases. Report of clinical statistics of Japan (2) (in Japanese). Nihon Rinsho 59 [Suppl 8]:317–327
Desnick RJ, Allen KY, Desnick SJ, Raman MK, Bernlohr RW, Krivit W (1973) Fabry’s disease: Enzymatic diagnosis of hemizygotes and heterozygotes. a-Galactosidase activities in plasma, serum, urine and leukocytes. J Lab Clin Med 81:157–171
Kitagawa T, Ishige N, Suzuki K, Owada M, Ohashi T, Kobayashi M, Eto Y, Tanaka A, Mills K, Winchester B, Keutzer J (2005) Non-invasive screening method for Fabry disease by measuring globotriaosylceramide in whole urine samples using tandem mass spectrometry. Mol Genet Metab 85:196–202
Nakao S, Takenaka T, Maeda M, Kodama C, Tanaka A, Tahara A, Yoshida A, Kuriyama M, Hayashibe H, Sakuraba H, Tanaka H (1995) An atypical variant of Fabry’s disease in men with left ventricular hypertrophy. N Engl J Med 333:288–293
Nakao S, Kodama C, Takenaka T, Tanaka A, Yasumoto Y, Yoshida A, Kanzaki T, Enriquez AL, Eng CM, Tanaka H, Tei C, Desnick RJ (2003) Fabry disease: Detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int 64:801–807
Desnick RJ, Banikazemi M, Wasserstein M (2002) Enzyme replacement therapy for Fabry disease, an inherited nephropathy. Clin Nephrol 57:1–8
Desnick RJ, Wasserstein MP, Banikazemi M (2001) Fabry disease (a-galactosidase A deficiency): renal involvement and enzyme replacement therapy. Contrib Nephrol 136:174–192
Ries M, Ramaswami U, Parini R, Lindblad B, Whybra C, Willers I, Gal A, Bech M (2003) The early clinical phenotype of Fabry disease: a study on 35 European children and adolescents. Eur J Pediatr 162:767–772
Ries M, Gupta S, Moor DF, Sachdev V, Qurik JM, Murray GJ, Rosing DR, Robinson C, Schaefer E, Gal A, Dambrosia JM, Garman SC, Brady RO, Schiffmann R (2005) Pediatric Fabry disease. Pediatrics 115:344–355
Mayes JS, Scheerer JB, Sifers RN, Donaldson ML (1981) Differential assay for lysosomal a-galactosidases in human tissues and its application to Fabry’s disease. Clin Chim Acta 112:247–251
Thadhani R, Wolf M, West ML, Tonelli M, Ruthazer R, Pastores GM, Obrador GT (2002) Patients with Fabry disease on dialysis in the United States. Kidney Int 61:249–255
Ohashi T, Sakuma M, Kitagawa T, Suzuki K, Ishige N, Eto Y (2007) Influence of antibody formation on reduction of globotriaosylceramide (GL-3) in urine from Fabry patients during agalsidase beta therapy. Mol Genet Metab 92:271–273
Maack T, Johnson V, Kau ST, Figueiredo J, Sigulem D (1979) Renal filtration, transport, and metabolism of low-molecular-weight proteins: a review. Kidney Int 16:251–270
Vander Jagt DJ, Steinberg BR, Glew RH (1992) Comparison of urinary excretion of four lysosomal hydrolases in healthy elderly and young adults. Clin Chem Acta 210:47–54
Berty RM, Adler S, Basu A, Glew RH (1990) Effect of acid base changes on urinary hydrolases in Fabry’s disease after renal transplantation. J Lab Clin Med 115:696–703
Paigen K, Peterson J, Ward E (1984) A genetic component in human lysosomal enzyme excretion. Biochem Genet 22:517–527
Hein LK, Bawden M, Muller VJ, Sillence D, Hopwood JJ, Brooks DA (2004) a-L-Iduronidase premature stop codons and potential read through in mucopolysaccharidosis type 1 patients. J Mol Biol 338:453–462
Karageorgos L, Harmatz P, Simon J, Pollard A, Clements PR, Brooks DA, Hopwood JJ (2004) Mutational analysis of mucopolysaccharidosis type VI patients undergoing a trial of enzyme replacement therapy. Hum Mutat 23:229–233
Brooks DA (1997) Protein processing: a role in the pathophysiology of genetic disease. FEBS Lett 409:115–120
Bradford TM, Litjens T, Parkinson EJ, Hopwood JJ, Brooks DA (2002) Mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome): a Y210C mutation causes either altered protein handling or altered protein function of N-acetylgalactosamine 4-sulfatase at multiple points in the vacuolar network. Biochemistry 41:4962–4971
Linthorst GE, Vedder AC, Aerts JM, Hollak CEM (2005) Screening for Fabry disease using whole blood spots fails to identify one-third of female carriers. Clin Chem Acta 353:201–203
Lukacs Z, Keil A, Kohlschütter A, Beck M, Mengel E (2005) The ratio of a-galactosidase to b-glucuronidase activities in dried blood for the identification of female Fabry disease patients. J Inherit Metab Dis 28:803–805
Lukacs Z, Hastung R, Beck B, Keil A, Mengel E (2007) Direct comparison of enzyme measurements from dried blood and leukocytes from male and female Fabry disease patients. J Inherit Metab Dis 30:614
Nagao Y, Nakashima H, Fukuhara Y, Shimmoto M, Oshima A, Ikari Y, Mori Y, Sakuraba H, Suzuki Y (1991) Hypertrophic cardiomyopathy in late-onset variant of Fabry disease with high residual activity of alpha-galactosidase A. Clin Genet 39:233–237
Sachdev B, Takenaka T, Teraguchi H, Tei C, Lee P, McKenna WJ (2002) Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation 105:1407–1411
Acknowledgments
This work was supported, in part, by a grant for the Research on Measures for Intractable Disease, Japanese Ministry of Health, Welfare and Labor (2001–2007), and by a grant for the development of a screening method for lysosomal storage disorders, from Genzyme Corporation (2002–2007).
We thank the doctors and their associates who sent us the urine samples to measure urinary α-gal A protein, as well as GL-3, and who were known to us the final diagnosis of the subjects at high risk for FD. We thank Ms. Joan Keutzer of Genzyme Corp., Cambridge MA, USA, for providing rabbit anti-human α-gal A polyclonal antibody, anti-human α-gal A monoclonal antibody, and C-17GL-3. We thank Dr. Gabriel Symonds for his help with correcting the English in this paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kitagawa, T., Suzuki, K., Ishige, N. et al. Non-invasive high-risk screening for Fabry disease hemizygotes and heterozygotes. Pediatr Nephrol 23, 1461–1471 (2008). https://doi.org/10.1007/s00467-008-0846-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-008-0846-6