
Abstract
Familial hypercholesterolemia results from mutations in the low-density lipoprotein (LDL) receptor or apolipoprotein B genes. We have previously reported the identification of a Utah autosomal-dominant hypercholesterolemia pedigree (kindred 1173) that did not show linkage to either of these loci (Hunt et al. 2000). Expansion of the pedigree and increased marker density within the region of interest have resulted in a multipoint LOD score of 9.6 and enabled us to decrease the size of the linked region to approximately 7.5 Mbp. In addition, we were able to identify additional families sharing the same microsatellite haplotype. While all haplotype carriers in kindred 1173 (K1173) are affected, the haplotype carriers within the newly identified families are unaffected, suggesting that the causal mutation in K1173 had occurred after divergence of these pedigrees from a common ancestor. Mutation screening of genes in the region identified a single nucleotide variant (G→T) present on the K1173 haplotype that was not present on the same haplotype in the other kindreds. This variant results in a D374Y missense change in the gene PCSK9.
Similar content being viewed by others
Introduction
Clinical familial hypercholesterolemia (FH) is characterized by LDL cholesterol levels of at least twice normal, with a bimodal inheritance pattern within affected families. In addition to dyslipidemia, affected individuals often present with tendon xanthomas and premature coronary disease. Mutations in either the LDL receptor gene (familial hypercholesterolemia, or FH) or the apolipoprotein B gene (familial defective apo B, or FDB) have been found to cause this disorder; however, these mutations explain only about 70% of all cases of the disease. A third locus (FH3) with a LOD score of 2.88 on chromosome 1p32 was initially reported by Varret et al. (1999), based on analysis of several French and Spanish FH families. Hunt et al. (2000), whose analysis of a large Utah FH pedigree identified a linkage at the same approximate location with a LOD score of 6.8, subsequently confirmed this linkage.
Recently, Abifadel et al. (2003) reported the results of mutation screening in the FH3 region. They identified two mutations in the gene PCSK9 (F216L and S127R), observed in three families with autosomal-dominant hypercholesterolemia. These mutations explained 12.5% of the hypercholesterolemia families examined in their study. Here we describe detection of another mutation in PCSK9 and propose that it is the causal mutation underlying our FH3 linkage.
Materials and methods
Pedigree ascertainment and description
Ascertainment of kindred 1173 (K1173) has been previously described by Haddad et al. (1999) and Hunt et al. (2000). The kindred was expanded to 95 individuals during the course of this study.
Genotyping
A genome search was performed on 31 members of K1173 as described by Hunt et al. (2000). Additional markers were genotyped on the expanded pedigree within a 17-cM region centered on the peak LOD score. The majority of markers are tri- or tetranucleotide repeats derived from public databases. Additional markers were developed by searching the public sequence databases for short tandem repeats within this region; these markers are indicated by the prefix MYR (sequence available upon request). The relative order of the markers within this interval was determined by use of the UCSC Human Genome Project Working Draft (http://genome.ucsc.edu; International Human Genome Sequencing Consortium 2001).
Linkage analysis
Linkage analysis was performed as described by Hunt et al. (2000).
Gene identification and assembly
Genes and EST clusters were identified using the UCSC Human Genome Project Working Draft (http://genome.ucsc.edu). Copy DNA sequences of incomplete genes were extended into their respective 5′UTRs by biotin capture 5′RACE (Tavtigian et al. 1996).
Mutation screening
Mutation screening was performed as described by Tavtigian et al. (1996).
Results
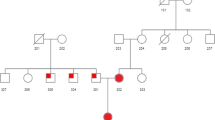
K1173 has been previously described in Haddad et al. (1999) and Hunt et al. (2000). During the course of this study K1173 has been expanded to a total of 95 individuals, including 32 affected individuals. All affected individuals had total cholesterol values at or above the 95th percentile for their age (Fig. 1a).

Pedigree drawing of K1173 showing phenotypes of family members. Only haplotype carriers and their spouses are shown. The shading of the symbols represent the following: black affected individuals, grey individuals of unknown phenotype, white unaffected individuals; * individual on statin treatment at the time of cholesterol measurement, and with historical total cholesterol levels >11.6 mmol/l, ** individuals with tendon xanthomas, statin treatment and historically high total cholesterol levels. Numbers under the symbols represent: top line individual identifier; middle line current age; bottom line highest recorded total cholesterol value (mmol/l). b Multipoint LOD scores for the linked region using a dominant model. Markers are listed in Table 1. Recombinant individuals in K1173 are indicated by the arrows below the LOD plot labeled with generation and individual number. Solid arrows depict haplotype sharing by affected individuals, dashed arrows indicate exclusion of the same region by unaffected family members. The dashed box outlines the minimal linked region as defined by recombinants from K1173. The location of PCSK9 is indicated by a vertical arrow
Hunt et al. (2000) reported a genome search for K1173 performed using 583 genetic markers with an average spacing of 5.7 cM. Multipoint linkage analysis using a dominant model identified a highly significant LOD score (6.8) on chromosome 1p32. In this study we have typed a total of 52 markers spanning this 17-cM linked region on all sampled individuals from K1173. Results of multipoint linkage analysis performed using MCLINK (Thomas et al. 2000) with a dominant model are shown in Fig. 1b. This analysis produced an extremely significant LOD score of 9.6 with very well defined recombinant boundaries.
All 32 affected individuals in the pedigree shared a single STR-based haplotype from marker 1-MYR0194 to marker 1-MYR0213 (Fig. 1 and Table 1). No unaffected pedigree members inherited this portion of the segregating haplotype, suggesting that the mutation in this pedigree is 100% penetrant. A total of 13 recombinant individuals were identified, ten affected and three unaffected. All of the recombinants were internally consistent and were used to define a 7.5-Mbp region that was likely to contain the disease-causing gene (Fig. 1b). Table 1 lists the markers, the chromosomal location and the multipoint LOD score at each marker in K1173.
We attempted to expand K1173 further by searching for the K1173 shared STR-based haplotype in other Utah dyslipidemia pedigrees that had been genotyped during our original genome search. Three such kindreds were identified; however, none of the individuals sharing the haplotype in these kindreds were affected. Table 1 shows the region shared in these kindreds (K1103, K602, and K3317) compared with the haplotype carried by affected individuals in K1173.
All genes identified within the minimal recombinant region were mutation screened on haplotype carriers from K1173 and K3317. A single nucleotide exchange (G→T) was found on the disease haplotype in K1173 that was not present on the same haplotype in K3317, this was the only observed difference between the shared haplotype carriers from the two pedigrees. The T allele was not detected in K1106 or K602, and 338 control chromosomes were all found to be homozygous for the wild-type G (data not shown). The mutation results in a missense change of D374Y in the gene PCSK9.
PCSK9 is a member of the peptidase S8 family of subtilases (serine proteases). The subtilase superfamily of serine proteases can be grouped into six families (A–F) based on sequence homology (Siezen and Leunissen 1997). PCSK9 shows greatest homology with the C family of serine proteases, which includes proteinase K. Comparison of PCSK9 to other family members indicates that while D374 is not a highly conserved residue it is located within a region of conservation in this family, and the most common residue at this location is D or G.
Discussion
Expansion of K1173, along with increased marker density, has narrowed the linked region to approximately 7.5 Mbp in size. We were also able to identify three distantly related pedigrees (K1103, K602 and K3317) with individuals that share the same STR-based haplotype but who are not affected. The identification of these pedigrees, in combination with the observation that the mutation in K1173 is 100% penetrant, led us to hypothesize that the mutation in K1173 is a recent event. Therefore, K1103, K602 and K3317 served as controls for mutation screening of the genes within the region. The causal mutation could be expected to reside on the haplotype in K1173 but be absent from the same haplotype in K1103, K602 and K3317.
Mutation screening of the coding exons within the region identified a single variant, a missense mutation in the gene PCSK9 (D374Y), located on the disease-associated haplotype in K1173 that was absent from the same haplotype in the other three kindreds. PCSK9 is a recently discovered member of a family of mammalian secretory kexin-like subtilases (Seidah et al. 2003). Members of this family are involved in protein activation, inactivation, and regulation of cellular location. Another family member, SKI-1 is involved in processing sterol regulatory element binding proteins (SREBPs) and hence plays an important role in cholesterol homeostasis. In a recent study, Maxwell et al. (2003) demonstrated that PCSK9 expression is downregulated in mice fed a high cholesterol diet, and that its expression is regulated by SREBPs. The catalytic domain of these enzymes, which is primarily responsible for substrate specificity, includes a triad of active residues comprised of Asp, His and Ser.
The Grantham chemical difference matrix (Grantham 1974) is a formula for calculating chemical difference values for amino acid substitutions that takes into account composition, polarity, and molecular volume. The chemical difference score correlates with the likelihood of an amino acid substitution affecting protein functioning. Recent studies propose that the average deleterious mutation has a chemical difference score of 93; in contrast, the average neutral substitution has a score of 63 (Miller and Kumar 2001; Abkevich V, Zharkikh A, Deffenbaugh AM, Frank D, Chen Y, Shattuck D, Gutin A, Tavtigian SV, manuscript submitted). The three mutations observed to date in PCSK9 have chemical difference scores of 22 (F216L), 110 (S127R), and 160 (D374Y). The mutation in K1173 shows 100% penetrance with affected individuals being diagnosed as young as one year old with a severe clinical phenotype (average untreated total cholesterol value in affected individuals of 9 mmol/l in K1173). The high chemical difference score for the D374Y mutation is consistent with the severity and penetrance of the mutation observed in K1173.
The identification of a causal mutation in PCSK9 resulting in hypercholesterolemia in a Utah pedigree substantiates the previously published work of Abifadel et al. (2003). This gene underlies a novel mechanism contributing to dyslipidemia, and it will be important to identify PCSK9’s substrate(s) and the impact of the mutations on its functioning. In addition, screening for variation in other hypercholesterolemia families will determine what fraction of disease families can be explained by mutations in the PCSK9 locus.
References
Abifadel M, Varret M, Rabes J-P, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, Derré A, Villéger L, Farnier M, Beucler I, Bruckert E, Chambaz J, Chanu B, Lecerf J-M, Luc G, Moulin P, Weissenbach J, Prat A, Krempt M, Junien C, Seidah NG, Boileau C (2003) Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 34:154–156
Grantham R (1974) Amino acid difference formula to help explain protein evolution. Science 185:862–864
Haddad L, Day INM, Hunt S, Williams RR, Humphries SE, Hopkins PN (1999) Evidence for a third genetic locus causing autosomal-dominant hypercholesterolemia: a non-LDLR, non-APOB kindred. J Lipid Res 40:1-10
Hunt SC, Hopkins PN, Bulka K, McDermott MT, Thorne TL, Wardell BB, Bowen BR, Ballinger DG, Skolnick MH, Samuels ME (2000) Genetic localization to chromosome 1p32 of the third locus for familial hypercholesterolemia in a Utah kindred. Artherioscler Thromb Vasc Biol 20:1089–1093
International Human Genome Sequencing Consortium (2001) Initial sequencing and analysis of the human genome. Nature 409:860–921
Maxwell KN, Soccio RE, Duncan EM, Sehayek E, Breslow JL (2003) Novel putative SREBP and LXR target genes identified by microarray analysis in liver of cholesterol-fed mice. J Lipid Res 44:2109–2119
Miller MP, Kumar S (2001) Understanding human disease mutations through the use of interspecific genetic variation. Hum Mol Genet 10:2319–2328
Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, Basak A, Prat A, Chrétien M (2003) The secretary proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci USA 100:928–933
Siezen RJ, Leunissen JAM (1997) Subtilases: the superfamily of subtilisin-like serine proteases. Protein Sci 6:501–523
Tavtigian SV, Simard J, Rommens J, Couch F, Shattuck-Eidens D, Neuhausen S, Merajver S, Thorlacius S, Offit K, Stoppa-Lyonnet D, Belanger C, Bell R, Berry S, Bogden R, Chen Q, Davis T, Dumont M, Frye C, Hattier T, Jammulapati S, Janecki T, Jiang P, Kehrer R, Leblanc JF, Goldgar D, et al. (1996) The complete BRCA2 gene and mutations in chromosome 13q-linked kindreds. Nat Genet 12:333–337
Thomas A, Gutin A, Abkevich V, Bansal A (2000) Multilocus linkage analysis by blocked Gibbs sampling. Stat Comput 10:259–269
Varret M, Rabes J-P, Saint-Jore B, Cenarro A, Marinoni JC, Civeira F, Devillers M, Krempf M, Coulon M, Thiart R, Kotze MJ, Schmidt H, Buzzi JC, Kostner GM, Bertolini S, Pocovi M, Rosa A, Farnier M, Martinez M, Junien C, Boileau C (1999) A third major locus for autosomal dominant hypercholesterolemia maps to 1p34.1-p32. Am J Hum Genet 64:1378–1387
Acknowledgements
This study was funded in part by Myriad Genetics, and National Institutes of Health grants HL47561 and HL21088. We would like to thank Lily L. Wu and Yuanpei Xin for expert technical assistance, and Georges Frech, Kirill Ostanin, and Steve Stone for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Timms, K.M., Wagner, S., Samuels, M.E. et al. A mutation in PCSK9 causing autosomal-dominant hypercholesterolemia in a Utah pedigree. Hum Genet 114, 349–353 (2004). https://doi.org/10.1007/s00439-003-1071-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-003-1071-9