Article Text

Abstract
Background Paroxysmal kinesigenic choreoathetosis (PKC) is characterised by recurrent and brief attacks of involuntary movement, inherited as an autosomal dominant trait with incomplete penetrance. A PKC locus has been previously mapped to the pericentromeric region of chromosome 16 (16p11.2-q12.1), but the causative gene remains unidentified.
Methods/results Deep sequencing of this 30 Mb region enriched with array capture in five affected individuals from four Chinese PKC families detected two heterozygous PRRT2 insertions (c.369dupG and c.649dupC), producing frameshifts and premature stop codons (p.S124VfsX10 and p.R217PfsX8, respectively) in two different families. Sanger sequencing confirmed these two mutations and revealed a missense PRRT2 mutation (c.859G→A, p.A287T) in one of the two remaining families. This study also sequenced PRRT2 in 29 sporadic cases affected with PKC and identified mutations in 10 cases, including six with the c.649dupC mutation. Most variants were truncating mutations, consistent with loss-of-function and haploinsufficiency.
Conclusion The present study identifies PRRT2 as the gene mutated in a subset of PKC, and suggests that PKC is genetically heterogeneous.
- Paroxysmal kinesigenic choreoathetosis
- targeted genomic sequencing
- PRRT2 mutations
- mutations
- complex traits
- epilepsy and seizures
- clinical genetics
- molecular genetics
- movement disorders (other than parkinsons)
- neurosciences
- nutrition and metabolism
- genetics
- oncology
- liver disease
- cancer: gastric
- linkage
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Paroxysmal kinesigenic choreoathetosis
- targeted genomic sequencing
- PRRT2 mutations
- mutations
- complex traits
- epilepsy and seizures
- clinical genetics
- molecular genetics
- movement disorders (other than parkinsons)
- neurosciences
- nutrition and metabolism
- genetics
- oncology
- liver disease
- cancer: gastric
- linkage
Introduction
Paroxysmal kinesigenic choreoathetosis (PKC, OMIM 128200) is the most frequently described type of paroxysmal dyskinesias disorder and is characterised by recurrent and brief attacks of involuntary movement.1 Familial and sporadic cases have been described. Familial PKC usually shows an autosomal dominant inheritance pattern with incomplete penetrance.
We previously performed linkage and haplotype analysis in four Chinese families (family 2 and family 4 with incomplete penetrance) with similar choreoathetosis clinical symptoms, and all mapped the disease locus to a region between D16S3093 and D16S3057 at 16p11.2-q12.1.2 3 This PKC critical region was in accordance with other studies.4 5 However, the causative gene remains unidentified. Recently, next generation sequencing has proven to be effective for discovering novel causal mutations in inherited diseases.
Methods
We sequenced the targeted region of linkage (chr16: 27 958 387–57 529 851), containing 237 RefSeq genes, in five individuals with PKC and one healthy control (did not carry the risk haplotype we previously identified) from four families in our previous report linked to this locus. The shotgun fragment libraries were hybridised to a custom NimbleGen array targeting the region and the eluate sequenced on the Illumina HiSeq 2000 platform. On average, 3.0 gigabases of data were generated per sample, to achieve at least 150-fold coverage of the mappable region (13 Mb) (details in supplementary table 1). We focused primarily on non-synonymous variants, splice site acceptor or donor variants, and coding insertions or deletions (NS/SS/I) that were most likely to be pathogenic (supplementary table 2).
Results
A total of 149–160 NS/SS/I variants were detected in 55 genes per affected individual. After filtering for variants found in dbSNP129, 1000 Genome Project and the healthy control sequencing data to remove shared variants, the number of variants was reduced approximately eightfold. A 0.60 Mb de novo microdeletion at 16p11.2 was identified in two sporadic PKC case reports,6 7 which suggested loss-of-function mutations seemed to cause the disorder and narrowed down the candidate region. Only one gene located in this region of the microdeletion, PRRT2, which encodes proline-rich transmembrane protein 2, harboured two heterozygous frameshift insertions in two families: a cytosine insertion (c.649dupC) in family 2 causing a frameshift and premature stop codon (p.R217PfsX8); and a guanine insertion (c.369dupG) in family 4 causing a frameshift and premature stop codon (p.S124VfsX10). In each family, the independent heterozygous mutations were indeed present in all available affected individuals and healthy risk haplotype carries we previously identified using Sanger sequencing.
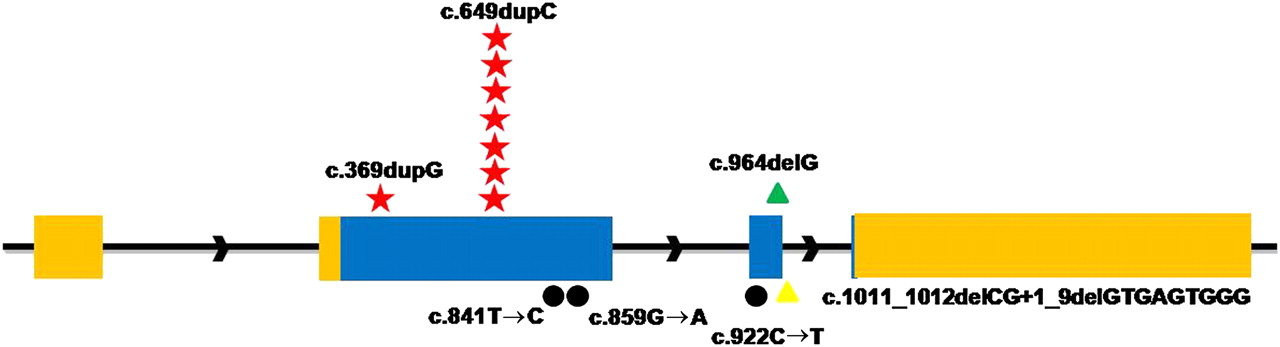
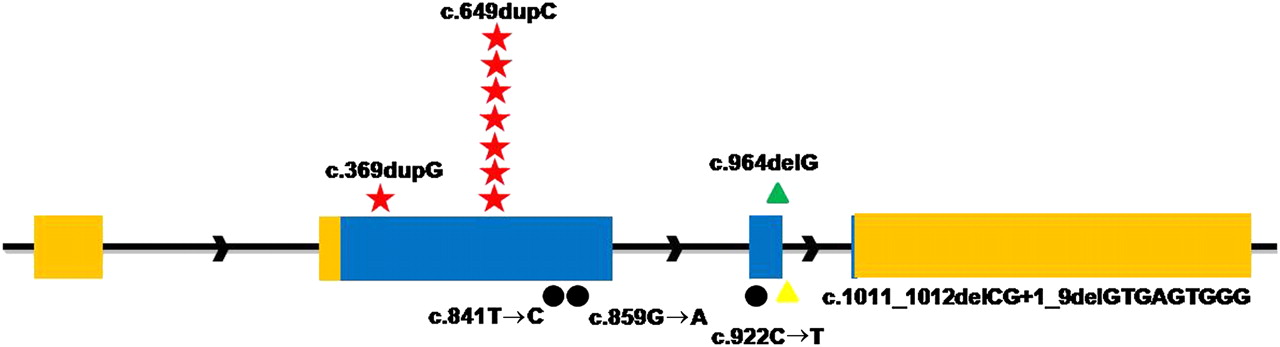
To confirm that mutations in PRRT2 were responsible for PKC, we screened the whole sequences of PRRT2 gene (exon, intron, 500 bp upstream of 5′UTR and 500 bp downstream of 3′UTR) in two other families (targeted genomic sequencing did not detect mutations likely due to the quality of sample) and 29 new PKC individuals. In family 1 (one of the two remaining families), a missense mutation (c.859G→A, p.A287T) in PRRT2 completely cosegregated with PKC phenotype, and five different mutations were detected in 10 sporadic PKC individuals. Collectively, seven different mutations were identified in PRRT2 by a combination of targeted genomic and Sanger sequencing (table 1, figure 1). None of these mutations was found in chromosomes from 192 ethnically matched control individuals.
Mutations in the PRRT2 gene by Sanger sequencing
{kind=link}
Identified mutations in the PRRT2 gene. Relative positions of mutations are indicated by symbols. Red Stars, frameshift insertions; green triangles, frameshift deletions; black dots, missense; yellow triangles, splice site mutations.
Discussion
Our results strongly suggest that mutations in PRRT2 are a cause of PKC. Little is known about PRRT2 or the functions of its encoded protein. In UniProtKB database, PRRT2 (Q7Z6L0) is predicted to be a multi-pass membrane protein consisting of an N-terminal extracellular domain containing a proline-rich domain and an N-glycosylation site, two transmembrane domains, and a C-terminal cytoplasmic domain. The N-terminal extracellular domain of PRRT2 is similar to PRiMA (proline-rich membrane anchor), which targets acetylcholinesterase to membrane.8 In addition, PRRT2 is also highly expressed in brain (Unigene). Therefore, we speculate that PRRT2 may serve as an anchor to connect with certain molecular at synapse.
In our Chinese PKC cohort, heterozygous mutations in PRRT2 explain 39.4% (13/33) of cases. Most of the mutations were predicted to truncate the polypeptide. One mutation was recurrent—c.649dupC—which was present in one family and six sporadic cases. Haploinsufficiency seems to be the most likely mechanism. Microdeletions of chromosome 16p11.2, encompassing PRRT2, have been reported in two sporadic PKC individuals.6 7 Also, three pathogenic missense mutations identified in family 1 and two sporadic cases were present at a conserved site among different species (supplementary figure 1). Different types of mutations may be associated with different phenotype. Clinical characteristics should be defined strictly. However, for one family (family 3) linked to the pericentromeric region of chromosome 16 and 19 sporadic cases, we screened the whole sequences of the PRRT2 gene and did not identify any mutations. It is possible that exonic deletions in PRRT2 or other mutations in distal gene regulatory sequences, or an additional causative gene on the same genomic region, are responsible for PKC. Alternatively, PKC appears to be genetically heterogeneous,9 10 and further analysis of these cases by exome sequencing may find additional genes.
Interestingly, PKC, benign familial infantile convulsions (BFIC2, OMIM 605751), infantile seizures and choreoathetosis (ICCA, OMIM 602066), and rolandic epilepsy with paroxysmal exercise induced dystonia and writer's cramp (RE-PED-WC, OMIM 608105) overlap across a pericentromeric region of chromosome 16,11 suggesting that they may be allelic disorders caused by PRRT2 mutations. Further functional studies of the PRRT2 gene and these specific gene mutations are needed to provide important insights into the pathophysiology of PKC and other movement disorders.
Acknowledgments
We are grateful to all of the subjects who kindly agreed to participate in this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (XLS) - Manuscript file of format xls
Footnotes
JL and XZ contributed equally to this work.
Funding This work was strongly supported by Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT IRT1006), the Science Fund for Creative Research Groups (30721063) and Natural Science Foundation of Beijing (7042037).
Competing interests None.
Patient consent Obtained.
Ethics approval This study was approved by the Ethics Committee of the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences.
Provenance and peer review Not commissioned; externally peer reviewed.