
Abstract
We describe two patients from distinct Cowden disease families with specific germline PTEN mutations whose disease differs from the usual appearance of Cowden disease. Their phenotype associates classical manifestations of Cowden disease and congenital dysmorphisms including segmental overgrowth, arteriovenous and lymphatic vascular malformations, lipomatosis and linear epidermal nevus reminiscent of the diagnosis of Proteus syndrome. We provide evidence in one of the two patients of a secondary molecular event: a loss of the PTEN wild-type allele, restricted to the atypical lesions that may explain an overgrowth of the affected tissues and the atypical phenotype. These data provide a new demonstration of the Happle hypothesis to explain some segmental exacerbation of autosomal-dominant disorders. They also show that a bi-allelic inactivation of PTEN can lead to developmental anomalies instead of malignant transformation, thus raising the question of the limitations of the tumor suppressive function in this gene. Finally, we suggest using the term ‘SOLAMEN syndrome’ (Segmental Overgrowth, Lipomatosis, Arteriovenous Malformation and Epidermal Nevus) in these peculiar situations to help the difficult distinction between the phenotype of our patients and Proteus syndrome.
Similar content being viewed by others

Introduction
The tumor-suppressor gene PTEN shows somatic mutations in a wide variety of sporadic neoplasias whereas its heterozygous germline mutations are responsible for a cancer-prone syndrome, Cowden disease (MIM 158350), and a related entity known as Bannayan-Riley-Ruvalcaba syndrome (MIM 153480). These diseases are characterized by a progressive development of numerous hypertrophic, hamartomatous and neoplastic lesions involving the skin, mucous membranes, gastrointestinal tract, thyroid, breast, brain and genitor-urinary system. Lipomas, angiomas and macrocephaly are frequently associated. The Proteus syndrome (MIM 176920), characterized by asymmetric and disproportionate overgrowth, cerebriform connective tissue nevi, the presence of an epidermal nevus and dysregulated adipose tissue, was attributed to PTEN germline mutations by one team who referred to this entity as Proteus or Proteus-like syndromes.1 However, that finding was not confirmed by others2 who questioned the accuracy of the patients' phenotypic evaluations and suggested misdiagnoses of Proteus syndrome.3
We describe two patients belonging to distinct Cowden disease families each associated with a germline PTEN mutation whose phenotypes mimicked Proteus syndrome. A secondary molecular event in one patient, a mosaic PTEN wild-type allelic loss, might explain the atypical phenotype.
Materials and methods
Report of cases
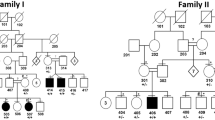
In the first family, the proband (Figure 1a: II9), a 30-year-old woman, had normal skin at birth but 5 days later a lymphatic vascular malformation appeared on her legs, back and abdomen. One month later, a difference in the length of her legs became evident. At 3 years of age, physical examination revealed macrocephaly, a few subcutaneous soft masses on the chest, overgrowth of the left thigh with a large vascular malformation on its anterior face and several lymphatic vascular malformations. At 7 years, four voluminous lipomas infiltrating the chest wall and one in the xyphoid region were surgically removed. At 8 years, two large lipomas in the left femoral triangle and the left thigh under a lymphatic vascular malformation were debulked. At 14 years, a microvesicular thyroid adenoma was removed. At 23 years, a mucinous cystadenoma of the left ovary was excised. At 26 years, the patient suffered a spontaneous fracture of the left hip. She underwent hip replacement and a bulky fibrous angiolipoma under the gluteus maximus was removed during the operation. Two years later, skin examination revealed two small gingival papules and large patchy lymphatic vascular malformations on the upper right and lower left parts of the back, the latter spreading to the left flank, hip and groin. An epidermal nevus following Blaschko's lines on the middle and upper left portion of the back extended to the left shoulder (Figure 2a). Marked hypertrophy of both calves was associated with right talipes varus limiting the patient's mobility. Subcutaneous varicosities and capillaries were present on the right thigh (Figure 2b), leg and sole (Figure 2c), whereas the big toe and medial ball of the left foot were ballooned and covered with several fibrotic papules (Figure 2d). Radiographs of the legs showed enlarged and dystrophic bones, diffuse demineralization and soft tissue calcifications in the right thigh. At 30 years, a diffuse fibrotic lipoma infiltrating the whole right calf was surgically removed in order to correct pes varus. Thyroid ultrasonography revealed multiple nodules and mammography detected fibrocystic breasts.

Pedigree of the two families reported. (a) and (b) The respective pedigrees of families 1 and 2. Gray symbols indicate the patients with Cowden disease and solid symbols the patients with Cowden disease mimicking Proteus syndrome. (c) carriers of the germline PTEN mutation; a transition A>G at nucleotide 403 leading to the change of the isoleucine 135 in valine for family 1 and a deletion of one nucleotide within a stretch of adenine (487–491) leading to a frameshift with a premature stop codon at 166 in family 2.

Cutaneous lesions of patient 1. An epidermal nevus (arrow) following Blaschko's lines extends from the upper left part of the back to the shoulder (a). Hypertrophy of the right thigh is associated with subcutaneous varicosities (b), deformation of the knee and a ‘hillocky’ appearance of the sole (c), whereas the medial ball of the left foot is ballooned (d) and covered with several fibrotic papules (arrows).
The other affected members of family 1 (Figure 1a: I1, II2, II6, II8, III4 and III6) presented a classical Cowden disease phenotype according to International Consortium criteria.4
In the second family, the proband (Figure 1b: III3), a 45-year-old woman, had a lumbar angioma at birth. One month later, overgrowth of the back of her right hand began and subsequently progressed. At 6 years, asymmetric overgrowth of the left leg was visible. One year later, a tumor was surgically removed from her right breast. At 10 years, the extramedullary part of an extensive fibrotic lipoma in the right paravertebral groove, infiltrating T9–T12 and the thoracic spinal canal, was partially removed. Six months later, she developed flaccid paraplegia, followed by an intrathoracic angioma and angiomas of the C7 and T5 vertebrae.
At 35 years, a multiheteronodular goiter was excised. Physical examination revealed macrocephaly, overgrowth of the right hand covered by multiple papules (Figure 3a) with a voluminous palmar sclerosing fibroma (Figure 3b), a lipoma of the left groin, a subcutaneous palpable vascular malformation on the left side of the chest and mild left leg overgrowth. At 44 years, intrathoracic arteriovenous shunts responsible for high-output heart failure were embolized and excised.

Morphological appearance of the right hand of patient 2. Hypertrophy of the right hand with macrodactyly is associated with multiple fibrotic papules (a) and a voluminous palmar storiform collagenoma (b).
The other affected family 2 members (Figure 1b: I2, II2, II3 and II6) presented a classical Cowden disease phenotype.
Methods
Using standard techniques, genomic DNA was extracted from the blood leukocytes of 11 members of family 1 and three members of family 2 who gave their informed consent. DNA was also extracted from various tissue biopsies and cultured skin-lesion fibroblasts from the two families' probands (patients 1 and 2). Some lesioned skin biopsies were subjected to dispase–digestion cleavage to allow separate testing of the epidermal sheet and dermis. PTEN mutations were sought in germline and somatic DNA after PCR-based denaturating gradient gel electrophoresis (DGGE) and dye-terminator sequencing using ABI prism 377 DNA sequencer as described previously.5
Microsatellite typing to search for 10qLOH was performed as described previously using a colorimetric-based detection (NBT – X phosphate) method for revelation.6 Six CA repeats flanking the PTEN locus were tested. Only informative markers were used for LOH analysis, that is D10S1765, D10S541, and D10S1739. PCR was performed using 50 ng of DNA template purified from blood leukocytes, cutaneous fibroblastic cells in culture and several frozen lesional biopsies. All allelic typing were repeated at least twice.
Results
PTEN mutation search
A PTEN missense germline variant in exon 5 (c.403A>G; p.I135 V) was found in the seven affected members of family 1 including patient 1 (Figure 1a) and was absent in four unaffected individuals. Sequencing of DNA from several biopsied lesions of patient 1 confirmed this PTEN I135 V germline variant. However, for three different lesions, that is the cutaneous fibroma of the left big toe, the epidermal nevus of the shoulder and the lipoma of the right calf, sequencing showed a clear decrease in the wild type allele (Figure 4). Sequencing of DNA from several other tissues including cutaneous fibroblastic cells in culture did not reveal any similar pattern and showed the germline variant in a heterozygous state, as observed previously at the leukocyte level. No other PTEN mutation was detected by DGGE.

Sequencing of the PTEN exon 5 in patient 1. The A>G transition at 403 codon (mutation p.I135 V) is found in the heterozygous state (thick arrows) in cutaneous fibroblastic cells in culture (a). The same mutation with a tendency to homozygosity and decrease of the peak height of the wild type allele (thin arrows) is observed for DNA from the cutaneous fibroma of the left big toe (b), the epidermal nevus of the shoulder (c) and the invasive lipoma of the right calf (d). Left: forward strand; right: reverse strand.
Two affected members of family 2 including patient 2 (Figure 1b) had a PTEN germline frameshift mutation in exon 5 (c.491delA; p.K164RfsX2) that was absent in one unaffected individual. DNA from cutaneous biopsy and cultured skin-lesion fibroblasts in patient 2 also contained this mutation, but in the heterozygous state. No other mutation was found in these samples.
Search for allelic loss
Owing to the tendency to homozygosity observed in the three lesions in patient 1, a loss of heterozygosity (LOH) for three PTEN-flanking markers was sought in DNA extracted from various tissues from the first patient. A clear decrease in intensity was observed for one allele of each marker with the DNA originating from the cutaneous fibroma of the left big toe, the epidermal nevus and the fibrotic lipoma of the right calf, compared to that of the blood leukocytes (Figure 5b). In contrast, LOH was not found in DNA from normal skin, cultured skin-lesion fibroblasts, other skin lesions or ovarian cyst. Haplotype analysis showed that LOH involved the maternal allele, that is the wild-type allele (Figure 5a). Separate searches for LOH in the epidermal sheet and the dermis of the cutaneous biopsies of the three lesions showing LOH revealed conservation of heterozygosity in the epidermis but LOH in the dermis sample (Figure 5c).

Microsatellite typing around the PTEN locus (centromere – D10S1765 – PTEN – D10S541 – D10S1739 – telomere) showing loss of heterozygosity in tissue samples. (a): allelic typing of blood leukocyte DNA from the proband's mother (lane 1), proband's father (lane 2) and patient 1 (lane 3), showing the disease-linked allele (arrow) in family 1. (b): loss of the wild-type allele at the PTEN locus detected in patient 1's DNA extracted from three different lesions compared to blood leukocyte (lane 1); the cutaneous fibroma of the left big toe (lane 2); the epidermal nevus on the back (lane 3); and the fibrotic lipoma on the right calf (lane 4). (c) allelic typing of various components of the cutaneous biopsy of the lesion on the left big toe showing allelic loss restricted to the dermis: cultured fibroblasts (lane 1); and after dispase digestion of the sample, the epidermal sheet (lane 2), and the dermis (lane 3).
No evidence of LOH was found for DNA extracted from cutaneous fibroblast cells in culture and from a right hand lesion biopsy from patient 2.
Discussion
We describe two families that had 12 members with manifestations of Cowden disease. The diagnosis was confirmed by genetic analysis demonstrating a PTEN mutation in the nine affected patients for whom blood samples were available. For family 1, the p.I135 V PTEN variant had previously been identified in another family with Bannayan-Riley-Ruvalcaba syndrome.7 To our knowledge, the functional consequence of this variant is not known, but it is probably deleterious because missense mutations of amino acids 133 and 136 were previously found to be responsible for the loss of PTEN lipid-phosphatase activity.8 The cosegregation of the disease with the variant in the family 1 (Figure 1) is demonstrative of linkage and single nucleotide polymorphism has never been described in this codon despite extensive human PTEN exploration in recent years. The mutation observed in family 2 (c.491delA) was also reported in another Cowden disease family.9 It causes the synthesis of a non-functional truncated protein so both these mutations are very probably responsible of the disease.
Our patients 1 and 2 had an atypical Cowden disease phenotype that could evoke Proteus syndrome. Both women had complex congenital dysmorphisms, including overgrowth of the legs, lipomatosis, vascular malformations and, in patient 1, a linear epidermal nevus and ovarian cystadenoma. However, they strictly do not meet the revised diagnostic criteria for Proteus syndrome.10, 11, 12 The foot and hand hypertrophic malformations presented by both patients (Figure 2c, d and 3) are different from the cerebriform connective tissue nevi of the palm or sole observed in Proteus syndrome.12 On the other hand, several findings including macrocephaly, thyroid adenoma, fibrocystic breast, gingival papules for the patient 1, and macrocephaly, multinodular goiter, multiple breast tumors for the patient 2, are suggestive of the ordinary nonsegmental Cowden disease.
To explain these additional manifestations in our patients, we looked for a second molecular event at the PTEN locus in lesional tissues. Three lesions in patient 1 show a tendency to homozygosity for PTEN mutation. This finding was confirmed by a decrease in intensity of the wild type allele of PTEN flanking markers suggestive of deletion of all or part of the 10q chromosomal arm in a subset of cells of these lesions. Interestingly, the deleted allele in each lesion carries the wild type PTEN allele. Availability of samples did not allow us to perform FISH experiments to investigate the PTEN locus at the cellular level.
These data are reminiscent of two recently published observations. The first one described a patient with a ‘Proteus-like’ syndrome associating congenital hemihypertrophy of the right leg, macrocephaly, epidermal nevi arranged in whirls, multiple lipomas and massive arteriovenous malformations involving the muscles and bones of the right leg reported by Zhou et al.13 That patient harbored a germline PTEN mutation (R335X) and a second-hit PTEN mutation (R130X) on the opposite allele in lesioned tissues, for example an epidermal nevus, a lipoma and an arteriovenous malformation. The second described a 3-year-old boy reported as Proteus syndrome by Loffeld et al14 showing an extensive epidermal nevus, macrocephaly, vascular malformation affecting both lower limbs, asymmetric hypertrophy of a leg, localized macrodactyly, and abdominal lipoma. This patient has a missense mutation of PTEN (p.Y68D) inherited from his mother who shows features of Cowden syndrome, as do several members of her family. Sequence analysis of the PTEN exon 3 from DNA extracted from curettings of the epidermal nevus shows a profile of homozygosity for the pY68D mutation evoking a PTEN wild-type allele loss.
The molecular data of these patients including ours showed that a heterozygous germline PTEN mutation, which is usually causative of Cowden disease, associated with a mosaic inactivation of the wild-type PTEN allele, may be associated with multiple dysmorphisms suggestive of other diseases (Proteus syndrome, Klippel-Trenaunay syndrome). These different lesions could be explained by the bi-allelic PTEN inactivation and a complete loss of the PTEN function responsible for hypertrophy of the affected tissues. The lesions in patient 2 are also probably related to a complete PTEN inactivation in specific tissues, even though we were unable to demonstrate a second PTEN alteration probably because of the small number of available samples. We indeed only tested one biopsy of the pathological skin and as we experimented for the first patient several skin samples from different areas have to be tested to demonstrate mosaicism. On the other hand, other mechanisms of inactivation like epigenetic silencing or a transcriptional defect cannot be excluded.
The same phenomenon is well known as ‘Knudson's hypothesis’ in malignant tumors arising in cancer-prone syndromes for which extensive demonstrations of a second hit restricted to tumoral cells was obtained.15 A similar mechanism with a second hit occurring earlier during development was postulated for autosomal-dominant genodermatoses16 and such a phenomenon was recently confirmed for Hailey-Hailey disease.17 Cowden disease is another example in which a germline dominant mutation can be associated with mosaic inactivation of the wild-type allele, resulting in segmental exacerbations of the disease.
However, although Cowden disease is a cancer-prone syndrome and PTEN a tumor-suppressor gene in accordance with Knudson's model,18, 19 these three observations demonstrate that the biallelic inactivation of PTEN does not lead systematically to malignant transformation. As malignant complications of Cowden disease are mainly of epithelial origin (breast carcinoma, thyroid carcinoma, renal cell carcinoma, endometrial carcinoma), it is likely that bi-allelic PTEN inactivation has different consequences depending on the cell types affected. We detected allelic loss in the dermis of the skin biopsies, not in epidermis, and in adipose and vascular tissues of the right calf fibrotic lipoma (data not shown) of patient 1. Similarly, Zhou et al13 demonstrated mosaic mutation in biopsies of a lipoma and a vascular malformation tissue from their patient suggestive of a mesenchymal origin. Allelic loss was not seen in the cultured cutaneous fibroblasts of our first patient and may involve other dermal cell types like adipocytes and/or endothelial cells, in which, unlike epithelial cells, PTEN nullizygosity might not lead to malignant transformation. If this hypothesis is correct, it is surprising that segmental exacerbation of Cowden disease is a rare phenomenon although malignant transformation is a very frequent complication of it. Epithelial cells might be more subject to genetic instability than connective tissue cells, as the high frequency of carcinoma with regard to sarcoma seems to suggest.
The first report of a PTEN mutation in a patient with a Proteus-like phenotype13 incited others to search for germline mutations in this gene in Proteus syndrome patients. The results of those studies were contradictory. PTEN mutations were found in 5/14 patients diagnosed with a Proteus or Proteus-like syndrome1 and in an isolated case presenting the Proteus phenotype.20 Other authors were unable to confirm these findings because no PTEN mutation was detected in 33 patients with Proteus syndrome.11, 21, 22 These contradictory data have generated a heated debate on the reliability of the phenotypes reported.3, 11, 23, 24 Our two patients and the case reported by Loffeld et al14 rekindle this controversy because, unlike the previously published cases, they are members of typical Cowden disease families with several affected members carrying the causal PTEN mutation. Their disease manifestations confirm that some patients with an inherited PTEN mutation may develop atypical lesions, such as segmental overgrowth, vascular malformations, multiple lipomas and epidermal nevus mimicking Proteus syndrome.
Our patient 2, lacking epidermal nevus indicates that such a phenotype can also show an incomplete presentation. In spite of obvious clinical overlap between these entities, some clinical findings should make it possible to distinguish them (Table 1). Epidermal nevus is common to the two diseases, whereas invasive lipomatosis and shunting arteriovenous malformation seem more specific of segmental exacerbation of Cowden disease. Conversely, cerebriform connective tissue nevus and disproportionate overgrowth with hyperostoses are more specific of Proteus syndrome.12, 25 In the event of segmental exacerbation of Cowden disease, the lesions observed are thickening of the sole as in our patient 1 and segmental proportionate overgrowth like the macrodactyly observed in our patient 2. Early manifestations such as macrocephaly should also point towards the diagnosis of a segmental form of Cowden disease. To clinically better distinguish between Proteus syndrome and segmental exacerbation of Cowden disease, we propose naming this latter entity the SOLAMEN syndrome with regard to the main lesions observed, that is Segmental Overgrowth, Lipomatosis, Arteriovenous Malformation and Epidermal Nevus.
References
Zhou XP, Hampel H, Thiele H et al: Association of germline mutation in the PTEN tumour suppressor gene and Proteus and Proteus-like syndromes. Lancet 2001; 358: 210–211.
Biesecker LG : The multifaceted challenges of Proteus syndrome. JAMA 2001; 285: 2240–2243.
Cohen MM, Turner JT, Biesecker L : Proteus syndrome: misdiagnosis with PTEN mutations. Am J Med Genet 2003; 122A: 323–324.
Eng C : Will the real Cowden syndrome please stand up: revised diagnostic criteria. J Med Genet 2000; 7: 828–830.
Lauge A, Lefebvre C, Laurent-Puig P et al: No evidence for germline PTEN mutations in families with breast and brain tumours. Int J Cancer 1999; 84: 216–219.
Dorion-Bonnet F, Mautalen S, Hostein I, Longy M : Allelic imbalance study of 16q in human primary breast carcinomas using microsatellite markers. Genes Chromosomes Cancer 1995; 14: 171–181.
Marsh DJ, Kum JB, Lunetta KL et al: PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet 1999; 8: 1461–1472.
Han SY, Kato H, Kato S et al: Functional evaluation of PTEN missense mutations using in vitro phosphoinositide phosphatase assay. Cancer Res 2000; 60: 3147–3151.
Eng C : PTEN: one gene, many syndromes. Hum Mut 2003; 2: 183–198.
Biesecker LG, Happle R, Mulliken JB et al: Proteus syndrome: diagnostic criteria, differential diagnosis and patient evaluation. Am J Med Genet 1999; 84: 389–395.
Biesecker LG, Rosenberg MJ, Vacha S, Turner JT, Cohen MM : PTEN mutations and Proteus syndrome. Lancet 2001; 358: 2079.
Turner JT, Cohen Jr MM, Biesecker LG : Reassessment of the Proteus syndrome literature: application of diagnostic criteria to published cases. Am J Med Genet 2004; 130A: 111–122.
Zhou XP, Marsh DJ, Hampel H, Mulliken JB, Gimm O, Eng C : Germline and germline mosaic PTEN mutations associated with a Proteus-like syndrome of hemihypertrophy, lower limb asymmetry, arteriovenous malformations and lipomatosis. Hum Mol Genet 2000; 9: 765–768.
Loffeld A, McLellan NJ, Cole T, Payne SJ, Ficker D, Moss C : Epidermal naevus in Proteus syndrome showing loss of heterozygosity for an inherited PTEN mutation. Br J Dermatol 2006; 154: 1194–1198.
Baker SJ, Kinzler KW, Vogelstein B : Knudson's hypothesis and the TP53 revolution. Genes Chromosomes Cancer 2003; 38: 329.
Happle R : Segmental forms of autosomal dominant skin disorders: different types of severity reflect different states of zygosity. Am J Med Genet 1996; 66: 241–242.
Poblete-Gutierrez P, Wiederholt T, König A et al: Allelic loss underlies type 2 segmental Hailey-Hailey disease, providing molecular confirmation of a novel genetic concept. J Clin Invest 2004; 114: 1467–1474.
Di Cristofano A, Pandolfi PP : The multiple roles of PTEN in tumor suppression. Cell 2000; 100: 387–390.
Sulis ML, Parsons R : PTEN: from pathology to biology. Trends Cell Biol 2003; 13: 478–483.
Smith JM, Kirk EP, Theodosopoulos G et al: Germline mutation of the tumour suppressor PTEN in Proteus syndrome. J Med Genet 2002; 39: 937–940.
Barker K, Martinez A, Wang R et al: PTEN mutations are uncommon in Proteus syndrome. J Med Genet 2001; 38: 480–481.
Thiffault I, Schwartz CE, Der Kaloustian V, Foulkes WD : Mutation analysis of the tumor suppressor PTEN and the glypican 3 (GPC3) gene in patients diagnosed with Proteus syndrome. Am J Med Genet 2004; 130A: 123–127.
Eng C, Thiele H, Zhou XP, Gorlin RJ, Hennekam RM, Winter RM : PTEN mutations and Proteus syndrome. Lancet 2001; 358: 2079–2080.
Pilarski R, Eng C : Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J Med Genet 2004; 41: 323–326.
Twede JV, Turner JT, Biesecker LG, Darling TM : Evolution of skin lesions in Proteus syndrome. J Am Acad Dermatol 2005; 52: 834–838.
Acknowledgements
We are grateful to our patients and their families who participated in this study, and to the clinicians who care for these patients. We thank Bernadette Gastaldello and Delphine Lafon for technical assistance, and Jean-Luc Birac for his help in preparing the figures. This study was partially funded by the Comités Dordogne et Charente Maritime de la Ligue Nationale Contre le Cancer.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Caux, F., Plauchu, H., Chibon, F. et al. Segmental overgrowth, lipomatosis, arteriovenous malformation and epidermal nevus (SOLAMEN) syndrome is related to mosaic PTEN nullizygosity. Eur J Hum Genet 15, 767–773 (2007). https://doi.org/10.1038/sj.ejhg.5201823
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201823
Keywords
This article is cited by
-
Diagnosis of PTEN mosaicism: the relevance of additional tumor DNA sequencing. A case report and review of the literature
BMC Medical Genomics (2023)
-
Genomic profiling informs diagnoses and treatment in vascular anomalies
Nature Medicine (2023)
-
Mosaic PTEN alteration in the neural crest during embryogenesis results in multiple nervous system hamartomas
Acta Neuropathologica Communications (2019)
-
Translation deregulation in human disease
Nature Reviews Molecular Cell Biology (2018)
