
Abstract
This study was undertaken to investigate the prevalence of BRCA1 and BRCA2 germline mutations in 91 German patients unselected for family history, who were diagnosed with breast cancer before the age of 41 years. Clinical information and blood samples were obtained from all patients. A comprehensive BRCA1 and BRCA2 mutational analysis was performed using the protein truncation assay and single-strand conformational polymorphism analysis followed by DNA sequencing of variant signals detected by these assays. Five different deleterious germline mutations including four frameshift mutations and one missense mutation were identified, three in BRCA1 (3.3%) and two mutations (2.2%) in BRCA2. Both BRCA2 mutations are novel and might be specific for the German population. An additional BRCA1 missense mutation previously described and classified as an unknown variant was found. This mutation was also detected in two breast cancer patients of family P 328 and not in 140 healthy controls suggesting that it is disease associated. In addition, one common polymorphism and five novel intronic sequence variants with unknown significance were found. Our findings show that mutations in BRCA1 and BRCA2 may contribute similarly to early-onset breast cancer in Germany. Given current constraints on health-care resources, these results support the notion that BRCA1 and BRCA2 mutation screening may have the strongest impact on health-care when targeted to high-risk populations.
Similar content being viewed by others


Main
Approximately 5% of breast and ovarian cancers are because of highly penetrant germline mutations in cancer predisposing genes. Two genes, BRCA11 and BRCA2,2,3 account for at least half of these cases.4 Individuals with mutations in these genes show increased lifetime risks of female breast cancer and ovarian cancer, and also prostate cancer and cancers at various other sites.5 In Germany, most information on the prevalence of BRCA1 and BRCA2 mutations has come from research on high-risk families selected for multiple occurrences of breast and ovarian cancer in multiple generations.6,7,8,9,10 Little is known about the prevalence of these mutations in other patient populations. Two previous studies analysing the prevalence of BRCA1 and BRCA2 mutations in German women with early-onset breast cancer (aged younger or equal to 35 and 40 years of age, respectively) have estimated a mutation frequency of 8% for BRCA1 and somewhere between 4 and 12.5% for BRCA2.10,11 Both studies, however, have been limited by a small number of cases (40 and 45, respectively), and only one focused on BRCA2.
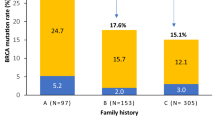
In this study, we searched for germline mutations in BRCA1 and BRCA2 in 91 women unselected for family history who were diagnosed with primary invasive or in situ breast cancer before the age of 41 years at the Städtisches Klinikum Karlsruhe between August 1993 and November 2001 and at the Women's Clinic, University of Heidelberg between July 1992 and April 1994 and September 1997 to August 1999, and with at least one parent with German ancestry. The mean age at onset of the disease of the study participants was 35 years (range 21–40 years). Written informed consent was obtained from all study participants. Of these patients, 53 were previously included in a screen for the 6 kb exon 13 duplication of the BRCA1 gene.12 Cancer diagnosis for each patient was confirmed by pathology reports in all cases. Family histories obtained from medical records and/or self-reported questionnaires were available from 73 cases. A family history was defined by at least one first- or second-degree female relative with breast or ovarian cancer. In all, 50 patients had no family history, 23 patients had a family history and no information was available from the remaining 18 patients. The clinical and pathological features of the study participants are shown in Table 1.
The complete coding regions of the BRCA1 and BRCA2 genes were screened as described previously.7,8,9 Genomic DNA from peripheral blood lymphocytes was used as the source of DNA for gene analysis. BRCA1 exon 11 and BRCA2 exons 10 and 11 were screened using the protein truncation test (PTT), and the remaining exons were screened by single-strand conformational polymorphism analysis (SSCP). Samples revealing variant bands were analysed using direct DNA sequencing. Two by two contingency tables were tested using Fisher's exact test. Two-sided P-values of 0.05 or less were considered significant.
Five different deleterious germline mutations were identified (5/91, 5.5%; 95% CI 1.8–12.4): three in BRCA1 (3/91, 3.3%; 95% CI 0.7–9.3) and two in BRCA2 (2/91, 2.2%; 95% CI 0.3-7.7) (Table 2a, b). Both BRCA2 mutations were novel and have not been previously described in other populations. They were not detected in 60 controls. The mean ages at diagnosis of breast cancer in BRCA1 mutation carriers (n=3) and BRCA2 mutation carriers (n=2) were 36 and 38.5 years, respectively. The mutations included four frameshift mutations predicted to cause premature termination codons at positions 1163 and 1829 of BRCA1 and at codons 1200 and 1229 of BRCA2 and one BRCA1 missense mutation predicted to destroy the protein RING-finger.
Five additional rare BRCA1 sequence variants were also identified: one missense mutation in exon 8 and four novel sequence variants in introns 13, 20, 22 and 24 (Table 2c). One sequence variant may be of functional importance. The missense mutation, Y179C, previously reported to the Breast Cancer Information Core database (BIC)13, affects the tyrosine residue at codon 179 that is conserved among mouse, dog and rat homologues.14,15 This mutation was also identified in the German breast cancer family P 328 (U Hamann, unpublished data). In this family, two females diagnosed with breast cancer at the ages of 51 and 67 years harboured the mutation, whereas one female diagnosed with breast cancer at the age of 34 years did not. It was not detected in 140 healthy controls. In BRCA2, a common K3326X nonsense mutation in exon 27 with uncertain significance and a novel nucleotide change in intron 26 were identified (Table 2d). All intronic BRCA1 and BRCA2 changes were assessed for their potential to affect RNA splicing using the algorithm available at the University of California, Berkley web site (http://www.fruitfly.org/seq_tools/splice.html). None was deemed to affect splicing efficiency.
Among the five women with deleterious BRCA mutations, all three women harbouring a BRCA1 mutation had a strong family history of breast cancer with at least two breast cancer cases among first- and second-degree relatives (Table 2a). The mother of patient P 465 was diagnosed with breast cancer at the age of 33 years and the paternal grandmother with bilateral disease at the ages of 41 and 68 years. As parental blood samples were available from the parents, the mutation was shown to be transmitted from the father. In the family of patient R 1018, the mother and a maternal aunt were diagnosed with breast cancer at the ages of 25 and 54 years, respectively. In this family the mutation most likely was inherited from the deceased mother, as the father did not carry the mutation. The mutation was also not detected in the affected aunt, indicating that she is a sporadic case. In the family of patient R 1198, the mother, a maternal aunt and the maternal grandmother were diagnosed with breast cancer at the ages of 39, 40 and 55 years, respectively. Patient R 423 carrying a BRCA2 mutation had no family history of breast/ovarian cancer (Table 2b). She was diagnosed with ovarian cancer at the age of 40 years, 1 year after the diagnosis of breast cancer.
In this study, similar mutation frequencies of 3.3% for BRCA1 and 2.2% for BRCA2 (p=1) were found in German breast cancer patients diagnosed before the age of 41 years and unselected for family history of the disease. Our mutation frequencies are comparable with those previously reported in other populations without strong founder effects. The mutation frequencies in various population-based studies in Britain, Australia, and North America were 3.5% in 254 cases under 3616, 4.1% in 57 cases under 3517, and 3.8% in 91 cases under 4018 for BRCA1 and 2.7% in 73 cases under 3319, and 2.4% in 254 cases under 36 for BRCA2.16 Some other studies estimated higher mutation frequencies ranging from 5.9 to 7.5% in 234 cases under 41 from Sweden20 and in 80, 193 and 203 cases under 35 from North America21,22,23 for BRCA1 and from 8.3% to 12.5% in 57 cases under 35 from Britain17 and in 40 cases under 40 from Germany11 for BRCA2. The different mutation frequencies within and between various populations may be explained by differences in the selection criteria, the sensitivity of the mutation detection or both. Further, estimates may be imprecise because of the small number of mutation carriers. However, it is also possible that these different frequencies may reflect real differences in the contribution of BRCA1 and BRCA2 mutations to early-onset breast cancer in populations that vary from one another in their racial and ethnic backgrounds.
The mutation frequencies observed in this study are likely to represent underestimates. Some of the BRCA1 sequence variants may be classified incorrectly as not disease-causing. In addition, we cannot exclude the possibility that some mutations have been missed by the mutation detection strategy adopted. Furthermore, the mutation frequencies may be higher as we did not screen for large genomic deletions and mutations in the promoter region.
Our findings show that mutations in the BRCA1 and BRCA2 genes may contribute similarly to early-onset breast cancer in Germany. Only a small proportion of early-onset breast cancer patients carry mutations in one or the other gene. Given current constraints on health-care resources, these results support the notion that BRCA1 and BRCA2 mutation screening may have the strongest impact on health-care when targeted to high-risk populations.
References
Miki Y, Swensen J, Shattuck-Eidens D et al: A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994; 266: 66–71.
Wooster R, Bignell G, Lancaster J et al: Identification of the breast cancer susceptibility gene BRCA2 [published erratum appears in Nature 1996; 379: 749]. Nature 1995; 378: 789–792.
Tavtigian SV, Simard J, Rommens J et al: The complete BRCA2 gene and mutations in chromosome 13q-linked kindreds [see comments]. Nat Genet 1996; 12: 333–337.
Ford D, Easton DF, Stratton M et al: Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. Am J Hum Genet 1998; 62: 676–689.
The Breast Cancer Linkage Consortium: Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst 1999; 91: 1310–1316.
Jandrig B, Grade K, Seitz S et al: BRCA1 mutations in German breast-cancer families. Int J Cancer 1996; 68: 188–192.
Hamann U, Brauch H, Garvin AM, Bastert G, Scott RJ : German family study on hereditary breast and/or ovarian cancer: germline mutation analysis of the BRCA1 gene. Genes Chrom Cancer 1997; 18: 126–132.
Hamann U, Haner M, Stosiek U, Bastert G, Scott RJ : Low frequency of BRCA1 germline mutations in 45 German breast/ovarian cancer families. J Med Genet 1997; 34: 884–888.
Hamann U, Liu X, Lange S, Ulmer HU, Benner A, Scott RJ : Contribution of BRCA2 germline mutations to hereditary breast/ovarian cancer in Germany. J Med Genet 2002; 39: e12.
Meindl A : Comprehensive analysis of 989 patients with breast or ovarian cancer provides BRCA1 and BRCA2 mutation profiles and frequencies for the German population. Int J Cancer 2002; 97: 472–480.
Plaschke J, Commer T, Jacobi C, Schackert HK, Chang-Claude J : BRCA2 germline mutations among early onset breast cancer patients unselected for family history of the disease. J Med Genet 2000; 37: e17.
The BRCA1 Exon 13 Duplication Screening Group: The exon 13 duplication in the BRCA1 gene is a founder mutation present in geographically diverse populations. Am J Hum Genet 2000; 67: 207–212.
Breast Cancer Information Core (BIC) database [http://www.nhgri.nih.gov/Intramural_research/Lab_transfer/Bic/]. 2002.
Chen KS, Shepel LA, Haag JD, Heil GM, Gould MN : Cloning, genetic mapping and expression studies of the rat Brca1 gene. Carcinogenesis 1996; 17: 1561–1566.
Szabo CI, Wagner LA, Francisco LV et al: Human, canine and murine BRCA1 genes: sequence comparison among species. Hum Mol Genet 1996; 5: 1289–1298.
Peto J, Collins N, Barfoot R et al: Prevalence of BRCA1 and BRCA2 gene mutations in patients with early-onset breast cancer. J Natl Cancer Inst 1999; 91: 943–949.
Anglian Breast Cancer Study Group: Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Br. J. Cancer 2000; 83: 1301–1308.
Southey MC, Tesoriero AA, Andersen CR et al: BRCA1 mutations and other sequence variants in a population-based sample of Australian women with breast cancer. Br J Cancer 1999; 79: 34–39.
Krainer M, Silva-Arrieta S, FitzGerald MG et al: Differential contributions of BRCA1 and BRCA2 to early-onset breast cancer. N Engl J Med 1997; 336: 1416–1421.
Loman N, Johannsson O, Kristoffersson U, Olsson H, Borg A : Family history of breast and ovarian cancers and BRCA1 and BRCA2 mutations in a population-based series of early-onset breast cancer. J Natl Cancer Inst 2001; 93:1215–1223.
Langston AA, Malone KE, Thompson JD, Daling JR, Ostrander EA : BRCA1 mutations in a population-based sample of young women with breast cancer. N Engl J Med 1996; 334: 137–142.
Malone KE, Daling JR, Thompson JD, O’Brien CA, Francisco LV, Ostrander EA : BRCA1 mutations and breast cancer in the general population: analyses in women before age 35 years and in women before age 45 years with first-degree family history. JAMA 1998; 279: 922–929.
Malone KE, Daling JR, et al: Frequency of BRCA1/BRCA2 mutations in a population-based sample of young breast carcinoma cases. Cancer 2000; 88: 1393–1402.
Acknowledgements
We are grateful to all patients for their participation in this study. This study was in part supported by the Tumorzentrum Heidelberg/Mannheim D.10029190. We thank Rodney J Scott for a critical reading of the manuscript and Antje Seidel-Renkert, Michaela Schleicher and Michael Gilbert for expert technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hamann, U., Liu, X., Bungardt, N. et al. Similar contributions of BRCA1 and BRCA2 germline mutations to early-onset breast cancer in Germany. Eur J Hum Genet 11, 464–467 (2003). https://doi.org/10.1038/sj.ejhg.5200988
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200988
Keywords
This article is cited by
-
The intronic BRCA1 c.5407-25T>A variant causing partly skipping of exon 23—a likely pathogenic variant with reduced penetrance?
European Journal of Human Genetics (2020)
-
Prevalence of BRCA mutations among hereditary breast and/or ovarian cancer patients in Arab countries: systematic review and meta-analysis
BMC Cancer (2019)
-
Comprehensive analysis of BRCA1 and BRCA2 germline mutations in a large cohort of 5931 Chinese women with breast cancer
Breast Cancer Research and Treatment (2016)
-
The prevalence and spectrum of BRCA1 and BRCA2 mutations in Korean population: recent update of the Korean Hereditary Breast Cancer (KOHBRA) study
Breast Cancer Research and Treatment (2015)
-
Characteristics and spectrum of BRCA1 and BRCA2 mutations in 3,922 Korean patients with breast and ovarian cancer
Breast Cancer Research and Treatment (2012)
