
Abstract
Pseudoxanthoma elasticum (PXE) is a hereditary disease characterized by progressive dystrophic mineralization of the elastic fibres. PXE patients frequently present with skin lesions and visual acuity loss. Recently, we and others showed that PXE is caused by mutations in the ABCC6/MRP6 gene. However, the molecular pathology of PXE is complicated by yet unknown factors causing the variable clinical expression of the disease. In addition, the presence of ABCC6/MRP6 pseudogenes and multiple ABCC6/MRP6-associated deletions complicate interpretation of molecular genetic studies. In this study, we present the mutation spectrum of ABCC6/MRP6 in 59 PXE patients from the Netherlands. We detected 17 different mutations in 65 alleles. The majority of mutations occurred in the NBF1 (nucleotide binding fold) domain, in the eighth cytoplasmatic loop between the 15th and 16th transmembrane regions, and in NBF2 of the predicted ABCC6/MRP6 protein. The R1141X mutation was by far the most common mutation identified in 19 (32.2%) patients. The second most frequent mutation, an intragenic deletion from exon 23 to exon 29 in ABCC6/MRP6, was detected in 11 (18.6%) of the patients. Our data include 11 novel ABCC6/MRP6 mutations, as well as additional segregation data relevant to the molecular pathology of PXE in a limited number of patients and families. The consequences of our data for the molecular pathology of PXE are discussed.
Similar content being viewed by others

Introduction
Pseudoxanthoma elasticum (PXE) is a hereditary disorder of the connective tissue. The disease is characterized by dystrophic mineralization of elastic fibres of the skin, retina, and cardiovascular system.1,2,3 Patients frequently have dermal lesions, experience progressive loss of visual acuity, and are at increased risk for cardiovascular complications. The clinical expression of PXE in patients is highly variable,4,5 and the mode of inheritance of PXE are currently not completely understood. The majority of PXE patients are sporadic cases. In a large subset of families, PXE segregates in an autosomal recessive (ar) fashion.6 In a small number of families, autosomal dominant (ad) inheritance has been reported.6,7
We and others previously localized the PXE gene to chromosome 16p13.1,8,9 and found that mutations in the ABCC6/MRP6 (MRP6) gene are associated with all genetic forms of PXE.10,11,12 The ABCC6/MRP6 gene is a member of the ATP-binding cassette (ABC) family, and encodes a transport protein of 1503 amino acids.13,14 The gene contains Walker A and B motifs typical for ABC proteins and 17 transmembrane domains.15 High ABCC6/MRP6 mRNA expression levels were found in kidney and liver, while lower expression was found in tissues usually affected by the disease.10,16 Using monoclonal antibodies, we recently localized human ABCC6/MRP6 to the basolateral side of hepatocytes and the proximal tubules of kidney.17 Recently, Ilias et al (2002) found that glutathione conjugates, including leukotriene-C4 (LTC4) and N-ethylmaleimide S-glutathione (NEM-GS), are actively transported by human ABCC6/MRP6. In three ABCC6/MRP6 mutant forms, loss of ABCC6/MRP6 transport activity appears to be directly responsible for PXE.18
Interpretation of the results from mutational analysis of ABCC6/MRP6 is complicated by the presence of two ABCC6/MRP6 pseudogenes in the genome and the multiple presence of larger and smaller deletions spanning (part of) the gene.10,19,20,21,22 In addition, still unknown molecular or environmental factors that could influence the variable clinical expression of the disease in patients complicate the correct assessment of genotype–phenotype relations. In this study, we present and discuss the results of ABCC6/MRP6 mutation analysis in 59 PXE patients and families from The Netherlands.
Results
Summary of mutational screening
The ABCC6/MRP6 gene was screened in 59 patients from apparently unrelated Dutch patients and families with PXE. In 41.9% of the families, PXE segregated in a clearcut ar fashion. A large proportion of patients (27.9%) were sporadic cases. In 30.2% of the families, the segregation pattern was not clear or revealed a putative dominant inheritance pattern (A Plomp, personal communication). Using PCR, SSCP, and direct sequencing, our ABCC6/MRP6 mutation detection rate was 55.1% (total number of mutations found divided by the total number of alleles). We detected at least one disease-causing allele in 43 patients (72.9%). We found 17 different ABCC6/MRP6 mutations that were assigned to 65 alleles (Table 1). A variety of mutations were observed including nonsense, missense, putative splice site mutations as well as deletions and one insertion. In our patient cohort, mutant alleles occurred in all combinations including homozygous, heterozygous, compound heterozygous, and hemizygous forms. A total of 11 different mutations were apparently unique to our patient group and have not been described by others. Combining our data with those of the literature, we conclude that 57 different ABCC6/MRP6 mutations are now known to cause PXE (Table 2).
Mutation types
The mutation types found in this study are summarized in Table 1. We observed two distinct nonsense mutations, R1141X and Q749X in 24 out of 117 alleles (20.5%). R1141X occurred in 22/117 alleles (18.8%) and was found in a homozygous, heterozygous, or compound heterozygous form in 19 patients (32.2%). This mutation was the most frequent ABCC6/MRP6 mutation found in our patient cohort. The Q749X nonsense mutation occurred in only two PXE patients in heterozygous or compound heterozygous form.
We found eight different missense mutations (R765Q, R1114H, T1130M, R1221C, A1303P, G1302R, K1394N, R1459C) that occurred in various combinations in nine alleles of eight patients. In addition, we detected five different intragenic ABCC6/MRP6 deletions. The first one was a 22 base pair(bp) deletion at position 1944 of the cDNA in exon 16. This deletion occurred in heterozygous and compound heterozygous form in two patients, and results in a shorter mRNA chain, and the introduction of a stop codon at cDNA position 2064. The second one was a deletion of a T at cDNA position 3775 in exon 27, which results in a frameshift at codon 1259 and premature termination at codon 1272. This mutation was detected in five alleles of three patients. The third deletion was a deletion of a G at position 4182, the last nucleotide of codon 1394 in exon 29, which changes the codon for Leu at 1402 into a stop codon. This deletion occurred in six alleles of five patients. The last deletion we observed spans exons 23–29, which is predicted to result in a 405 amino-acid deletion in the polypeptide. The latter deletion was found on 13 alleles (11.1%) of 11 patients (18.6%) and was the second most frequent mutation in this study after R1141X.
One allele with a large deletion encompassing the entire ABCC6/MRP6, ABCC1 and MYH11 genes was found in one patient. Apart from the five intragenic deletions, a putative splice site deletion mutation (IVS17-12 delTT) was found. The only insertion detected was a 4 bp insertion at position 4220, codon 1047, which alters the reading-frame, and most likely abolishes protein function.
In 20 PXE patients, we only found a single ABCC6/MRP6 mutation in one allele, but no nonsynonymous sequence changes in the second allele.
Mutation spectrum and distribution
The summary of our data, and our data combined with those of the literature are presented in Figure 1 and Table 2. Our data indicate that the diversity among the ABCC6/MRP6 nonsense mutations and deletions in our patient cohort was relatively low. However, the few nonsense mutations, which we did identify, occurred relatively frequently. In contrast, the diversity among our missense mutations was large, while the frequency of each missense mutation was relatively low.

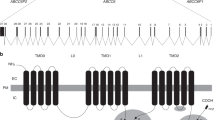
A schematic representation of the MRP6 protein with 17 transmembrane domains is shown. Note that the glycosylated NH2 terminus is located outside the cell. The carboxy terminus is located inside the cell. The two ATP-binding domains are indicated with two black circles. The mutation spectrum in ABCC6/MRP6 associated with PXE is shown. Novel mutations presented in this study, or previously reported by us, are marked with asterisks.
The mutations we found in our cohort occurred all in cytoplasmatic domains of the ABCC6/MRP6 toward the carboxy-terminal end of the protein, within or beyond the first NBF1 domain. We detected three clusters of mutations in the predicted ABCC6/MRP6 protein: in the NBF1 domain, in the 8th cytoplasmatic loop between the 15th and 16th transmembrane regions, and in NBF2 (see Table 1 and Figure 1). In the NBF1 domain, we found three different NBF1-specific mutations in five alleles (4.3%). In the second, cytoplasmatic-loop cluster of mutations, we detected mutations in 25 alleles (21.4%) of 21 patients (excluding large deletions spanning this domain). The R1141X mutation, the most common PXE mutation found in our cohort, is located in this domain. Five different NBF2-specific mutations were found on nine alleles (7.7%) from seven patients. Outside these three domains, relatively infrequent and widely distributed mutations are found corresponding to 11 alleles of nine PXE patients. Indeed, the mutation distribution in our cohort reflects those in other studies if we combine all ABCC6/MRP6 mutation data known to date: Approximately 80% of the mutations occur in cytoplasmatic domains; NBF1 is mutated on eight ABCC6/MRP6 alleles (2.3%), the eighth cytoplasmatic domain on 79 alleles (22.3%), and NBF2 on 41 alleles (11.6%).
Segregation of PXE in pedigrees
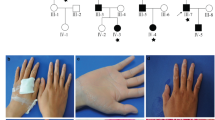
To illustrate the complex genetics of PXE, we studied a number of patients and pedigrees from our cohort in more detail (Figure 2). The clinical features of these patients are presented in Table 3.

Pedigrees of three Dutch families and haplotypes of microsatellite markers within or flanking the ABCC6/MRP6 gene. Black squares and circles denote severely affected individuals and half-black/half-white symbols mildly affected individuals. Exact description of the phenotypes are presented in Table 3. The bars denote haplotypes segregating with the PXE phenotype. The ABCC6/MRP6 gene is located in between the markers D16S764 and 118F2TAA; − indicates the absence of the mutation and + the presence of the mutation. In PXE patients of pedigree P 26095, detailed DNA and RNA analyses revealed one allele with a mutation (R1495C) as well as a wild-type allele. The R1495C mutation segregates through the maternal line, since a nephew of the mother carries the mutation also. Note that in pedigree P26098, for a number of markers (indicated with !!), no Mendelian inheritance is observed, which suggests the presence of a submicroscopic deletion. Indeed, further molecular analysis indicated that this deletion spans the ABCC6/MRP6, ABCC1, and MYH1 genes.
The PXE patient from pedigree PXE26101 was initially classified as a ‘sporadic’ patient. Molecular analysis revealed that she was homozygous for the ABCC6/MRP6 R1141X mutation. Both her healthy parents were carriers of this mutation, which is compatible with normal ar inheritance. Using RT-PCR and direct sequencing, we found that this mutation was also present in ABCC6/MRP6 mRNA isolated from the patients’ peripheral blood.
We previously reported on a sporadic patient with a large DNA deletion spanning the ABCC6/MRP6, ABCC1, and MYH11 genes on the maternal allele.10 We initially suggested that PXE in this patient (II-1 in pedigree PXE26098, also called P-06) could be caused by haploinsufficiency at the ABCC6/MRP6 locus, given the apparent absence of a mutation in the second allele. However, an intragenic deletion spanning exons 23–29 on the paternal allele initially escaped our attention. Molecular analysis of the breakpoints of this latter deletion showed that it was similar to the exons 23–29 deletion recently reported by Ringpfeil et al.21 This intragenic deletion in the family was confirmed to be present in mRNA from leukocytes. Consequently, PXE in this (compound heterozygous deletion) patient is most likely caused by complete loss of function of ABCC6/MRP6. It is remarkable that the patient (II-1) was severely affected, while his brother, who had the same genotype for the ABCC6/MRP6 locus, has only asymptomatic angioid streaks.
In a putative ad PXE family, molecular analysis was carried out (PXE 26095). The family consisted of an affected mother, a healthy father, three severely affected, two mildly affected, and three nonaffected offspring. The characteristics of the phenotype are given in Table 3. We detected an ABCC6/MRP6 missense mutation (R1459C) heterozygously present in the DNA and RNA of all affected individuals.
We were not able to find a mutation or deletion on the second allele in the patients’ DNA. Normal segregation and heterozygosity of multiple polymorphic markers intragenic and flanking ABCC6/MRP6 markers do not suggest the presence of a large deletion extending into ABCC6/MRP6 either. Restriction analysis of an RT-PCR product of exon 30 revealed the presence of the R1459C mutation in one transcript, while the other transcript was wild type (Figure 3). Sequencing of the entire ABCC6 cDNA in two patients (II-2, II-7) through RT-PCR of RNA from peripheral blood showed the presence of a mutated (R1459C), as well as an entirely normal, wild-type ABCC6 transcript, without mutations (not shown).

RT-PCR analysis of ABCC6 expression in leukcocytes in individuals homozygous and heterozygous for the R114X mutation and in individuals heterozygous for the R1459C mutation compared with wild-type ABCC6. RT-PCR primers and conditions are given in the Material and methods section. (a) A wild-type cDNA fragment (wt) from the RT-PCR reaction is cut with BsiY1 in fragments of 400 and 100 bp. The R1141X mutant allele leads to loss of a BsiYI restriction site; the mutated allele is therefore not cut and presents with a 500 bp band. (b) The R1495C mutation abolishes a AciI restriction site in the cDNA: Wild type sequences are cut and result in AciI fragments of 310 and 30 bp; R1495C mutated fragments result in a single AciI fragment of 340 bp. All findings (a and b) were confirmed by direct sequencing.
We found the R1459C mutation also in the DNA of a maternal nephew, who, unfortunately, refused clinical examination (not shown). However, the latter finding provides further evidence that the R1459C mutation in this pedigree segregates via the maternal line to the affected children. No consanguinity between the parents was found for four generations back.
Discussion
PXE mutations – Functional consequences
Although our mutation detection rate (55.1%) is comparable to another study of smaller European cohorts,22 it remains likely that we still did not detect a substantial amount of mutations. This is probably because of a number of reasons. First, it is well known that the sensitivity of SSCP is only 70–80%. Consequently, 20–30% of the mutations will not be detected. Second, a number of disease-causing mutations may still be present in those promotor or intron regions that we did not screen (yet). In addition, we may have missed mutations in the ABCC6/MRP6 promotor region given its homology with ABCC6/MRP6-ψ2 pseudogene sequences. Finally, we may have missed still intragenic deletions using Southern analysis because of crosshybridization with putative pseudogene sequences.
An alternative explanation of our results may be that other, not directly ABCC6/MRP6-related genes or genetic or environmental factors are involved in the expression of the PXE phenotype or that rare ABCC6/MRP6 mutations cause a dominant PXE phenotype. If the latter is the case, a yet-to-be defined number of individuals with an ABCC6/MRP6 mutation in a single allele, may actually present with (almost) the complete disease phenotype.
The distribution of PXE-associated mutations in ABCC6/MRP6 is unequal. The majority of the mutations occurred toward the carboxy-terminal end and in the cytoplasmatic domains of the protein. This phenomenon can be partly explained by the intracellular and carboxyterminal location of the evolutionary conserved NBF1 and NBF2 domains, known to be essential for ATP-driven transport of the ABC-protein family. Indeed, the clustering of mutations at the NBF1 and NBF2 domains of ABCC6/MRP6 indicates that these two regions are essential for the normal function of this transport protein. For NBF1, the latter was recently demonstrated by transport activity studies.18
In order to obtain further clues for gene function, we compared the (mutated) protein sequences of ABCC6/MRP6 with mutations and sequences found in other members of the ABC protein family. We compared sequences and mutations in ABCC6/MRP6 with ABCC2, ABCC8, ABCC7, and ABCR, which are implicated in, respectively, Dubin–Johnson syndrome, familial persistent hyperinsulinaemic hypoglycaemia of infancy, cystic fibrosis, and Stargardt disease or perhaps even age-related macula degeneration.23,24,25,26,27 As we expected, a number of similar mutations occurred in conserved regions of the ABC proteins or even in the same conserved residues. In all proteins, a large number of mutations implicated in disease occurred in the NBF1 and NBF2 domains. Further alignment showed that the R765Q mutation in ABCC6/MRP6 is the positional equivalent of both the R560T mutation in ABCC7,28 and the R842G mutation in ABCC8.29 Similarly, additional possible positional equivalent clusters of conserved and mutated residues were found between ABCC6/MRP6 and ABCC2 (R1114H and R1150H),30 ABCC6/MRP6 and ABCC7 (3775 del T and W1204X),31 ABCC6/MRP6 and ABCR (R1459C and H2128R, 4220InsAGAA and R2077W, R1141X and L1631P).32,33
Interestingly, for both ABCC7 and ABCR, models were postulated in which the severity of the disease shows an inverse correlation with the predicted transport activity of the ABC protein. According to these models, the mutation type (‘mild’ or ‘severe’) of mutations determines the transport activity of ABCC7 and ABCR34,35 and, consequently, the phenotype. On the basis of our data, we suggest that such a model is not directly applicable to ABCC6/MRP6 and PXE. We observe considerable clinical variability between PXE family members, who, obviously, carry the same mutation, and we suggest that the severity of the PXE phenotype is not directly correlated with the level of ABCC6/MRP6 activity.
Molecular pathology of PXE
The majority of mutations found in ABCC6/MRP6 were nonsense mutations, deletions, as well as missense mutations in conserved regions of the gene. These mutations are expected to result in either a shorter or dysfunctional mRNA or an absent or dysfunctional protein. The latter is most likely compatible with a complete loss of functional ABCC6/MRP6 protein in homozygote patients, and an ar inheritance pattern. Indeed, for three ABCC6/MRP6 mutations, Ilias et al18 recently found that loss of ABCC6/MRP6 transport activity was directly responsible for PXE.
As confirmed by our data and segregation studies, autosomal recessive segregation was observed in the majority of our PXE families, and may also explain a large part of the so-called sporadic cases. However, there are a number of reports in the literature of families in which PXE potentially segregates in an ad fashion.6,7 These segregation patterns may result from pseudodominant inheritance, as a result of parental consanguinity, or (mild) manifestation of the disease in heterozygotes.36,37,38,39 On the other hand, the presence of rare ABCC6/MRP6 mutations resulting in a true ad disease and inheritance pattern cannot be ruled out completely. In this study, we presented a novel family with an R1459C ABCC6/MRP6 mutation, in which ad segregation of PXE on the basis of clinical, molecular, and genealogical data, is the most likely explanation for our results.
Further studies using functional (transport) assays in patient fibroblasts, cell lines, or using transgenic mice technology are essential to elucidate the true consequences of the latter and other mutations. Extensive and careful clinical and molecular examination of additional individual PXE patients and pedigrees carrying unique mutations, will, no doubt, contribute to a more complete understanding of the molecular pathology of PXE.
Materials and methods
Clinical examination and human materials
We obtained permission from the Medical Ethical Committee of the Academic Medical Centre in Amsterdam for all studies with human subjects or human material. PXE patients were either ascertained through the national register of genetic eye disease at the Netherlands Ophthalmic Research Institute or through collaboration with physicians. All patients are of Dutch descent.
The diagnosis of PXE in individuals was based on the results of ophthalmological and dermatological examinations. In most patients, histopathological study was performed by skin biopsy, usually from affected skin in the neck, including a von Kossa staining.
Ophthalmological examination included visual acuity measurement, slit-lamp examination, ophthalmoscopy, and frequently fluorescein angiography. Cardiovascular examinations, if performed, included electrocardiograms (ECG). The diagnosis of PXE was considered if there were yellowish papules or plaques on the lateral side of the neck and/or flexural areas of the body, typical histopathological changes in skin biopsy and at least one of the following retinal abnormalities: peaud’ orange, angioid streaks, or comets (white punched-out lesions).
Molecular analysis
DNA was isolated from peripheral blood samples of PXE patients and their families according to standard protocols, essentially described elsewhere.40 Molecular analysis of the ABCC6 gene and FISH analysis were essentially carried out as described.10
PCR primers were selected from the published sequence of human chromosome 16 BAC clone A-962B4 (GenBank Accession No. U91318), TIGR database (http://www.tigr.org), or the primers were a gift of collaborators (C Boyd). To distinguish between MRP6 gene and pseudogene sequences, novel primers for exons 1–9 were used, as described elsewhere.20
To screen both exon and the adjacent intron sequences, PCR products were derived from intronic sequences 20–50 bp out from the end of each ABCC6/MRP6 exon. PCR was performed on DNA of each PXE patient and the products were analysed with SSCP. Fragments with a mobility shift were characterized by direct sequencing. Heterozygote detection on the ABI-310 was always checked manually by traditional Sanger dideoxy sequencing.
The potential presence of intragenic large deletions of genomic DNA was confirmed by consistent lack of amplification of the relevant exons in patients who were heterozygous or homozygous for deletions. Deletions of exons 15 and 23–29 were detected using primers and methods essentially described elsewhere.22
To identify additional putative mutations in patients where we could only identify one or no mutations, Southern blot analysis with ABCC6/MRP6 exons as a probe was carried out, essentially as described.10 The probes were hybridized against the corresponding genomic DNA of patients that was cut with at least two different restriction enzymes. In addition, we sequenced the promoter region (1 kb before the first ATG) in 14 patients and, finally, we sequenced the entire gene in nine patients. Haplotype analysis with microsatellite DNA markers was carried out in families as described previously.10 In addition, several new intragenic ABCC6 markers were used (Table 4).
The definition of disease-associated alleles essentially follows the criteria described by Le Saux et al.22 In summary, sequence variants predicted to result in nonsense or splice-site changes were considered to be disease-associated alleles if they are absent in DNA of a panel of at least 100 controls. Other variants, such as missense mutations, were considered to be disease associated if they were absent in the same control panel, segregated with the disease in pedigrees, and involve evolutionary conserved amino-acid residues.
Expression studies
Total RNA was isolated from blood and cultured skin fibroblasts from different individuals carrying different ABCC6/MRP6 mutations by use of RNAzol reagent (Biotech. Laboratory). First-strand cDNA was synthesized with Superscript reverse transcriptase (Life Technologies) and oligo T18 primers (Boehringer). cDNA aliquots were subjected to amplification with appropriate primers followed by direct sequencing.
The ABCC6/MRP6-specific RT-PCR primers used to analyse the mutations indicated were as follows: (R1141X): ABCC6/MRP6F, 5′-CTGTCTCCAAGCCATTGGGC-3′ (cDNA position 3008–3027) and ABCC6/MRP6R, 5′-AGCCACCAGTCGCGGGAAAC-3′ (cDNA position 3524–3505); (deletion exon 23–29): ABCC6/MRP6F3, 5′-ATACGGCAGGGTGAAGGCCA-3′ (cDNA position 2801–2820) and ABCC6/MRP6R3, 5′-CAGTGCACTGTGCAAACCAGC-3′ (cDNA position 4380–4360); (R1459C): ABCC6/MRP6F4, 5′-CTGGCTCTCTGCGGATGAAC-3′ (cDNA position 4081–4100); ABCC6/MRP6R4,5′-AGAACCCGGGCACAGTCCAT-3′ (cDNA position 4432-4413).
Database sequences and amino-acid sequence alignment
GenBank sequences NP001162 (ABCC6/MRP6), Q92878 (ABCC2), XP004980 (ABCC7) NP00343 (ABCC8), and NP000341 (ABCR) were used to construct an alignment of the ABCC protein sequences and analyse the evolutionary conservation of corresponding amino-acid positions. Sequences were aligned using CLUSTAL algorithm.
Accession codes
References
Goodman RM, Smith EW, Paton D et al: Pseudoxanthoma elasticum: a clinical and histopathological study. Medicine 1963; 42: 297–334.
Lebwohl M, Schwartz E, Lemlich G et al: Abnormalities of connective tissue components in lesional and non-lesional tissue of patients with pseudoxanthoma elasticum. Arch Dermatol 1993; 285: 121–126.
McKusick VA : Pseudoxanthoma elasticum, in Mckusick VA: Heritable disorders of connective tissue. St Louis: Mosby, 1972, pp 475–520.
Sherer DW, Bercovitch L, Lebwohl M : Pseudoxanthoma elasticum: significance of limited phenotypic expression in parents of affected offspring. J Am Acad Dermatol 2001; 44: 534–537.
Uitto J, Boyd C, Lebwohl M, Rosenbloom J, Terry S : International Centennial Meeting of Pseudoxanthoma Elasticum: progress in PXE research. J Invest Dermatol 1998; 110: 840–842.
Neldner KH : Pseudoxanthoma elasticum. Clin Derm 1988; 6: 83–92.
Hausser I : Anton-Lamprecht: Early preclinical diagnosis of dominant pseudoxanthoma elasticum by specific ultrastructural changes of dermal elastic and collagen tissue in a family at risk. Hum Genet 1991; 87: 693–700.
Struk B, Neldner KH, Rao VS, St Jean P, Lindpaintner K : Mapping of both autosomal recessive and dominant variants of pseudoxanthoma elasticum to chromosome 16p13.1. Hum Mol Genet 1997; 6: 1823–1828.
Van Soest S, Swart J, Tijmes N, Sandkuijl LD, Rommers J, Bergen AAB : A locus for autosomal recessive pseudoxanthoma elasticum, with penetrance of vascular symptoms in carriers, maps to chromosome 16p13.1. Genome Res 1997; 7: 830–834.
Bergen AAB, Plomp AS, Schuurman EJ et al: Mutations in ABCC6/MRP6 cause pseudoxanthoma elasticum. Nat genet 2000; 25: 228–231.
Le Saux O, Urban Z, Tschuch C et al: Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat Genet 2000; 25: 223–228.
Ringpfeil F, Lebwohl MG, Christiano AM, Uitto J : Pseudoxanthoma elasticum: mutations in the MRP6 gene encoding a transmembrane ATP-binding cassette (ABC) transporter. Proc Natl Acad Sci USA 2000; 97: 6001–6006.
Borst P, Evers R, Kool M, Wijnholds J : A family of drug tranporters: the multidrug resistance-associated proteins. J Natl Cancer Inst 2000; 92: 1295–1302.
Cole SP, Bhardwaj G, Gerlach JH et al: Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 1992; 258: 1650–1654.
Higgins CF . ABC transporters: from microorganisms to man. Annu Rev cell Biol 1992; 8: 67–113.
Kool M, van der Linden M, de Haas M, Baas F, Borst P : Expression of human MRP6, a homologue of the multidrug resistance protein gene MRP1: in tissues and cancer cells. Cancer Res 1999; 59: 175–182.
Scheffer GL, Hu X, Pijnenborg AC, Wijnholds J, Bergen AA, Scheper RJ : MRP6 (ABCC6/MRP6) detection in normal human tissues and tumors. Lab Invest 2002; 82(4): 515–518.
Ilias A, Urban Z, Seidl TL et al: Loss of ATP dependent transport activity in pseudoxanthoma elasticum associated mutants of ABCC6/MRP6 (MRP6). J Biol Chem 2002; 277(19): 16860–16867.
Cai L, Lumsden A, Guenther UP et al: A novel Q378X mutation exists in the transmembrane transporter protein ABCC6/MRP6 and its pseudogene: implications for mutation analysis in pseudoxanthoma elasticum. J Mol Med 2001; 79: 536–546.
Pulkkinen L, Nakano A, Ringpfeil F, Uitto J : Identification of ABCC6/MRP6 pseudogenes on human chromosome 16p: implications for mutation detection in pseudoxanthoma elasticum. Hum Genet 2001; 109: 356–365.
Ringpfeil F, Nakano A, Uitto J, Pulkkinen L : Compound heterozygosity for a recurrent 16.5-kb Alu-mediated deletion mutation and single-base-pair substitutions in the ABCC6/MRP6 gene results in pseudoxanthoma elasticum. Am J Hum Genet 2001; 68: 642–652.
Le Saux O, Beck K, Sachsinger C et al: A spectrum of ABCC6/MRP6 mutations is responsible for pseudoxanthoma elasticum. Am J Hum Genet 2001; 69: 749–764.
Paulusma CC, Bosma PJ, Zaman GJ et al: Congenital jaundice in rats with a mutation in a multidurg resistance-associated protein gene. Science 1996; 271: 1126–1128.
Riordan JR, Rommens JM, Kerem B et al: Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989; 245: 1066–1073.
Thomas PM, Cote GJ, Wohllk N et al: Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science 1995; 268: 426–429.
Toh S, Wada M, Uchiumi T et al: Genomic structure of the canalicular multispecific organic anion–transporter gene (MRP2/cMOAT) and mutations in the ATP-binding-cassette region in Dubin-Johnson syndrome. Am J Hum Genet 1999; 64: 739–746.
Allikmets R, Shroyer NF, Singh N et al: Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science 1997; 277: 1805–1807.
Kerem BS, Zielenski J, Markiewicz D et al: Identification of mutations in regions corresponding to the two putative nucleotide (ATP)-binding folds of the cystic fibrosis gene. Proc Natl Acad Sci U S A 1990; 87: 8447–8451.
Fournet JC, Mayaud C, de Lonlay P et al: Unbalanced expression of 11p15 imprinted genes in focal forms of congenital hyperinsulinism: association with a reduction to homozygo-sity of a mutation in ABCC8 or KCNJ11. Am J Pathol 2001; 158: 2177–2184.
Mor-Cohen R, Zivelin A, Rosenberg N, Shani M, Muallem S, Seligsohn U : Identification and functional analysis of two novel mutations in the multidrug resistance protein 2 gene in Israeli patients with Dubin–Johnson syndrome. J Biol Chem. 2001; 276: 36923–36930.
Ghanem N, Costes B, Girodon E, Martin J, Fanen P, Goossens M : Identification of eight mutations and three sequence variations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genomics 1994; 21: 434–436.
Maugeri A, van Driel MA, van de Pol DJ et al: The 2588G→C mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease. Am J Hum Genet 1999; 64: 1024–1035.
Fishman GA, Stone EM, Grover S, Derlacki DJ, Haines HL, Hockey RR : Variation of clinical expression in patients with Stargardt dystrophy and sequence variations in the ABCR gene. Arch Ophthalmol 1999; 117: 504–510.
Welsh MJ, Tsui L-C, Boat TF, Beaudet AL : Cystic fibrosis, in: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill, Inc., 1995, pp 3799–3876.
van Driel MA, Maugeri A, Klevering BJ, Hoyng CB, Cremers FP : ABCR unites what ophthalmologists divide(s). Ophthalmic Genet 1998; 19: 117–122.
Ringpfeil F, Pulkkinen L, Uitto J : Molecular genetics of pseudoxanthoma elasticum. Exp Dermatol 2001; 10: 221–228.
Bacchelli B, Quaglino D, Gheduzzi D et al: Identification of heterozygote carriers in families with a recessive form of pseudoxanthoma elasticum. Mod Pathol 1999; 12: 1112–1123.
Uitto J, Pulkkinen L, Ringpfeil F : Molecular genetics of pseudoxanthoma elasticum: a metabolic disorder at the environment–genome interface? Trends Mol Med 2001; 7: 13–17.
Struk B, Cai L, Zach S et al: Mutations of the gene encoding the transmembrane transporter protein ABC-C6 cause pseudoxanthoma elasticum. J Mol Med 2000; 78: 282–286.
Miller SA, Dykes DD, Polesky HF : A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
Meloni I, Rubegni P, De Aloe G et al: Point mutations in the ABCC6/MRP6 gene and a large deletion including also ABCC1 and MYH11. Hum Mutat 2001; 18: 85.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hu, X., Plomp, A., Wijnholds, J. et al. ABCC6/MRP6 mutations: further insight into the molecular pathology of pseudoxanthoma elasticum. Eur J Hum Genet 11, 215–224 (2003). https://doi.org/10.1038/sj.ejhg.5200953
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200953
Keywords
This article is cited by
-
Dietary magnesium, not calcium, prevents vascular calcification in a mouse model for pseudoxanthoma elasticum
Journal of Molecular Medicine (2010)
-
ABCC6 and pseudoxanthoma elasticum
Pflügers Archiv - European Journal of Physiology (2007)
-
Pseudoxanthoma Elasticum: the End of the Autosomal Dominant Segregation Myth
Journal of Investigative Dermatology (2006)
-
Pseudoxanthoma elasticum
Der Ophthalmologe (2006)
-
Analysis of ABCC6 (MRP6) in normal human tissues
Histochemistry and Cell Biology (2005)
