
Abstract
It has long been known that the salivary glands of hematophagous (blood-feeding) arthropods secrete soluble apyrases, which are potent nucleotide hydrolyzing enzymes capable of hydrolyzing extracellular ATP and ADP, the latter being a major agonist contributing to platelet aggregation. Only recently, however, has the identification of proteins homologous to these apyrases been reported in non-blood-feeding organisms such as rodents and humans. In this review, we present an overview of the diverse family of apyrases first described in the blood-feeding arthropods, including the identification and characterization of the soluble and membrane-bound vertebrate enzymes homologous to these arthropod apyrases. We also describe the enzymatic properties and nucleotide specificities of the expressed enzymes, and insights gained into the structure and function of this calcium activated nucleotidase (CAN) family from biophysical, mutagenesis and crystallography studies. The potential therapeutic value of these proteins is also discussed.
Similar content being viewed by others

Overview of the ‘soluble’ apyrase family
Hemostasis (the prevention of blood loss) is a vertebrate process with three main components: The plasma protein clotting factor cascade; vasoconstriction; and platelet aggregation. To facilitate feeding, hematophagus (blood-feeding) arthropods such as fleas [1], ticks [2], biting midges [3], and sand flies [4, 5] have evolved mechanisms to disarm all three components and thereby inhibit hemostasis in their hosts. Contained within the saliva of such blood-feeding organisms are a variety of potent bioactive compounds, including anticoagulants, vasodilators, and antiplatelet factors. For recent reviews on the antihemostatic strategies of blood-feeding arthropods, see [6–8].
We are particularly interested in one mechanism utilized by these blood-feeding organisms to inhibit hemostasis: The injection of soluble salivary apyrases (ATP and ADP hydrolyzing enzymes, EC 3.6.1.5) at the site of host skin puncture. These apyrases are members of a family of calcium-dependent proteins that rapidly hydrolyze extracellular nucleotides such as adenosine tri- and diphosphate [9]. Lacerations produced by the arthropod’s mouth parts whilst probing for a blood vessel in the host likely expose platelets to copious amounts of extracellular ADP, which is released from injured cells and from the dense granules of activated platelets. ADP represents one of the most important physiological agonists for platelet recruitment, aggregation and plug formation via activation of platelet purinergic receptors (recently reviewed in [10–13]), and the apyrases facilitate the blood-meal acquisition for these organisms by inhibiting the ADP-mediated activation of host platelets through an increase in ADP catabolism [8].
Although these soluble apyrases were initially described in the saliva of several different types of blood-feeding arthropods, the cloning of the Cimex lectularius (bedbug) apyrase resulted in the identification of homologous proteins in non-blood-feeding vertebrates (human and mouse) [9]. Although only identified through deduced amino acid sequences in the GenBank expressed sequence tags (ESTs) database, with no protein expression data to support their presumed apyrase activity, that report established the potential existence of proteins in non-blood-feeding vertebrates that are clearly related to the salivary apyrases found in blood-feeding invertebrates. Subsequent to that report, two studies describing the identification of very similar cDNAs from the blood-feeding arthropods Lutzomyia longipalpis (sandfly) and Phlebotomus papatasi (sandfly) were published [4, 5]. These studies showed the similarity in enzymatic function between the three blood-feeding arthropod enzymes, and pointed out additional putative apyrases in Caenorhabditis elegans and Drosophila melanogaster based on sequence homologies. The amino acid sequences of these putative Ca2+-dependent apyrases are conserved among arthropods and vertebrates (e.g., bedbug and human apyrases show 45% overall amino acid identity) and appear to have diverged from a common ancestral enzyme [9] (see Figure 1). Whether these vertebrate sequences encoded true apyrases with similar enzymatic properties and physiological functions however, would not be known until later.

Phylogenetic analysis of the human calcium activated nucleotidase and related family members. The human protein sequence, along with the 10 other most closely related sequences, were aligned using the computer program CLUSTALW, and phylogenetic distances were determined and presented graphically by the Phylip Drawtree program, available through the Biology Workbench of the San Diego Supercomputer Center at http://workbench.sdsc.edu/. The lengths of the lines connecting the proteins are proportional to the phylogenetic distances between the sequences. The GenBank accession numbers of the proteins in this work are given in parentheses: Human (Homo sapiens) (AAM94564), Mus musculus (AAH20003), Rattus norvegicus (NP_653355), Drosophila melanogaster (AAF54638), Cimex lectularius (AAD09177), Phlebotomus papatasi (AAG17637), Lutzomyia longipalpis (AAD33513), Anopheles gambiae (CAC35453), Xenopus laevis (AAH41215), Bos taurus (XP_596269), Gallus gallus (XP_427661).
Cloning, expression and enzymatic characterization of the vertebrate homologues of the arthropod salivary apyrases
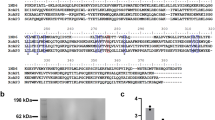
Further analysis of multiple sequence alignments of vertebrate and invertebrate family members by Failer et al. [14] revealed eight conserved clusters of amino acids presumably important for structure and enzymatic activity. No relatedness was found to the eNTPDase nucleotidase family of enzymes and their unique ‘apyrase conserved regions’ [15]. Consistent with their subcellular localization studies, the presence of an extended amino-terminus and RXR endoplasmic reticulum retention/retrieval motif was found in all three vertebrate sequences examined (rat, mouse, and human, see Figure 2). The rat N-terminal sequence is predicted to be a hydrophobic sequence that allows retention in the cellular membranes of transfected CHO cells. The authors concluded that N-terminal sequence shortening and signal peptide cleavage may be a specific evolutionary adaptation of the blood-feeding arthropod apyrases. It should be noted that the prediction as to whether or not there is a signal peptide cleavage site in these proteins (using the ‘SignalP’ program at the ExPASy (Expert Protein Analysis System) website: http://us.expasy.org/) depends on the assumed start of the amino acid sequences, which depends on which of the multiple possible start codons/initiation methionines are presumed to initiate translation, which, in most cases, is unknown.

Multiple sequence alignment of three vertebrate and three blood-feeding arthropod CAN nucleotidase family members. The full-length mouse, rat, and human protein sequences, along with three blood-feeding arthropod sequences (bed bug, and two different sand flies), were aligned using the computer program CLUSTALW alignment algorithm, available through the Biology Workbench of the San Diego Supercomputer Center at http://workbench.sdsc.edu/. Residues were shaded to indicate various levels of conservation using the BOXSHADE program, also available through the Biology Workbench toolbox. Those amino acids that are strictly conserved among all family members are indicated by black shading. Those amino acids that are similar at a given position are indicated by gray shading. The N-terminal RXR endoplasmic reticulum retention/retrieval motif of the vertebrate sequences first described by Failer et al. [14] is represented by a black bar. The key residues for nucleotide and calcium binding (determined through crystallography of the human CAN by Dai et al. [20]) are represented by pluses (+) and asterisks (*), respectively. The seven amino acids mutated by Yang and Kirley [18] are represented by: Open circles (○) for those four mutations having very little activity remaining; filled circles (≆) for those two mutations having substantial residual activity; and a triangle (≆r) for the mutation (Glu to Tyr) which increased ADPase nucleotidase activity approximately five-fold. The five point mutations that convert the wildtype human CAN into a 100 fold more potent ADPase capable of abolishing platelet aggregation are shown as carets beneath the sequence (^). The accession numbers of the sequences used in this alignment are listed in the legend to Figure 1.
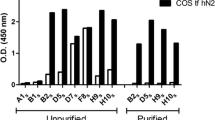
The same year that the characterization of the rat homologue was reported, Smith et al. described the cloning and expression of the human homologue of the bed bug apyrase (first referred to as SCAN-1, for soluble calcium activated nucleotidase-1 [16]). When expressed in COS-1 cells, many similarities were found to the rat nucleotidase, including the strict requirement for calcium for activity, a pH optimum around 7, and a nucleotide preference of UDP > GDP ≫ ADP, with no detectable hydrolysis of nucleoside monophosphates. Unlike the expressed rat enzyme however, which was shown to be membranebound [14], the human enzyme that was characterized was a soluble protein found in the conditioned media of transfected cells, likely produced by cleavage at the predicted signal peptide cleavage site (see Figure 2) [16].
Further enzymatic characterization of the human nucleotidase was performed by Murphy et al. [17]. Those investigators expressed the soluble portion of the human enzyme in E. coli, and refolded the enzyme from the resultant bacterial inclusion bodies. The bacterially expressed human apyrase was similar in enzymatic properties to the mammalian cell produced soluble form, indicating that a transmembrane domain and post-translational modifications that do not occur in bacteria (such as glycosylation) are not necessary for proper folding of the protein or nucleotidase activity. Many divalent cation chloride salts were tested (CaCl2, MgCl2, MnCl2, SrCl2, CuCl2, CoCl2, and NiCl2), but only calcium was able to support nucleotide hydrolysis. Interestingly, those authors also found that the human protein was a calcium-binding protein (as detected by changes in protein absorbance, fluorescence, and activity as a function of calcium concentration) with a calcium-triggered transition from an enzymatically inactive to an active form with halfsaturation at about 90 µM CaCl2 at low ionic strength [17].
Another important characteristic of the human calcium-activated nucleotidase that distinguishes it from other unrelated nucleotidases such as the naturally occurring soluble forms of human NTPDases (i.e., NTPDase5 and NTPDase6) is its remarkable stability. For example, Valenzuela et al. [5] reported that the P. papatasi apyrase was successfully renatured after SDS-PAGE, while the bed bug apyrase was resistant to denaturation by the 0.1% trifluoroacetic acid and acetonitrile used for purification by reverse-phase chromatography [9]. The wildtype human enzyme has been reported to be stable for over 24 h at 37°C in the presence of calcium, and for more than one week at room temperature [18]. Indeed, the enzymatic activity of this human CAN protein is retained for at least six months when stored in buffer at 4°C under sterile conditions (T.M. Smith, personal observations). Thus, the human calcium activated nucleotidase is considerably more stable than the naturally occurring soluble NTPDase apyrases, NTPDase5 and NTPDase6 (T.L. Kirley, personal observations). In fact, in stark contrast to the human CAN, purified NTPDase5 (also known as CD39L4) was noted to be unstable and prone to aggregation [19].
Mutagenesis and crystallographic analysis of the human calcium activated nucleotidase
Extensive mutagenesis studies have recently been performed on the human enzyme in an effort to evaluate key amino acids in this family [18, 20]. Yang and Kirley [18] used computational analysis of multiple sequence alignments of invertebrate and vertebrate members of the apyrase family to identify eight highly conserved regions of amino acids. The conserved regions were designated as nucleotidase conserved regions 1–8 (NCR 1–8) and they were used to direct a mutagenesis approach to identify amino acids important for enzymatic function. Four amino acids (Asp or Arg residues) contained in the NCRs were mutated to alanine and, consistent with their degree of conservation, all four mutations had substantial detrimental effects on the enzyme activity. Several of these mutants failed to bind the nucleotide analog Cibacron blue and were sensitive to limited tryptic digestion, suggesting alterations in their nucleotide binding pockets and/or their ability to bind calcium and undergo the calcium-induced conformational change. One mutation in the human enzyme was reported (E130Y), which was targeted for mutagenesis due to differences found between the sequences of the vertebrate and invertebrate sequences at that residue (Figure 2). This E130Y mutation increased GDPase activity two-fold and ADPase activity five-fold, which led the authors to suggest that this amino acid was important for determining the nucleotidase specificity for this family.
Interestingly, it was found that either CaCl2 or SrCl2 could induce the conformational change detected by the increases in tryptophan fluorescence, confer resistance to proteolysis, and provide thermal stability of the enzyme at 37°C, although only calcium could support nucleotide hydrolysis [18]. This suggested that calcium plays an important role not only as a nucleotide co-substrate, but also for maintaining the conformation and structural stability of the enzyme.
In addition to performing extensive mutagenesis studies on the human protein, Dai et al. [20] reported the highresolution crystal structures of the enzyme both in the presence and absence of the non-hydrolyzable GDP substrate analog GMP-CP. The structures revealed a novel nucleotide-binding motif comprising a five-blade beta propeller structure, with a single calcium-binding site in the middle of the central tunnel (Figure 3), coordinated by Ser 100, Glu 147, Glu 216, Ser 277, and Glu 328 (numbering based on the soluble version of the apyrase minus the signal peptide sequence). The single bound calcium ion also contacts a water molecule and the carboxylate group of Asp 101. With the exception of a conserved glutamate substitution in the blood-feeding arthropod proteins for Asp 101, the calcium binding residues are invariant in all members (Figure 2). Consistent with this observation, mutagenesis of these amino acids abolished the enzymatic activity. The presence of the substrate analog GMP-CP revealed that the guanine ring is bounded by Trp 165 and Tyr 239. In addition, the phosphate recognition pocket is lined mostly by hydrophilic residues, and site-directed mutagenesis of these amino acids confirmed their roles in the active site of the enzyme [20].

Structure of the human calcium activated nucleotidase in complex with the GMP-CP substrate analog. Ribbon diagrams of the human protein β-propeller structure as viewed along (A) or perpendicular to (B) the central tunnel. The polypeptide is colored from blue at the C-terminus through to red at the C-terminus. The structure reveals an unusual five-bladed β-propeller with five twisted β-sheets, each formed from four antiparallel β-strands. The interface between neighboring blades is predominantly hydrophobic, with residues on the adjacent faces of the β-sheets in van der Waals contact. The Ca2+ ion (green sphere) is located at the middle of the central tunnel. The GMP-CP molecule is shown in a ball-and-stick representation (CPK color scheme). The propeller has an approximate diameter of 44 Å and height of 30 Å. The atomic coordinates are available at the Protein Data Bank (PDB code 1S1D). Reprinted from Cell, Vol. 116, Dai et al., Structure and Protein Design of a Human Platelet Function Inhibitor, pp 649–659, Copyright (2004), with permission from Elsevier.
As mentioned earlier, the wild-type human enzyme is very unlike the arthropod enzymes regarding its ability to hydrolyze adenosine-based nucleotides, with the human enzyme preferentially hydrolyzing UDP and GDP nucleotides. In addition, the human enzyme had no effect on an in vitro assay of ADP-induced platelet aggregation, unlike the invertebrate enzymes. Indeed, the tick apyrase has even been reported to cause disaggregation and dispersal of ADP-aggregated platelets [21]. The crystallographic data of the human protein combined with sequence analyses of the blood-feeding arthropod apyrases suggested that substrate preference differences between the human and arthropod family members resulted from divergence at one or more of the non-conserved active site-residues. Thus, in an effort to make the human enzyme more like the blood-feeding arthropod counterparts with respect to preferentially hydrolyzing ADP and inhibiting platelet aggregation, Dai et al., 2004 [20] undertook a structureguided mutational analysis of the human enzyme to identify and incorporate residues which contribute to preference for ADP as substrate. 35 amino acid substitutions were made, with a combination of five of those mutations (see Figure 2) resulting in an engineered enzyme that had a Ka for calcium activation of ADP hydrolysis in the physiological range (dropping from 5.2 mM in the wild-type to 1.6 mM in this multiple mutant), and a 100 fold higher rate of ADP hydrolysis. This engineered G160S, L161S, K163M, T168K, E178M enzyme, designated HB (human/bedbug) apyrase, was also examined with respect to its ability to inhibit ADP and collagen mediated platelet aggregation. As shown by Dai et al. [20], this guided mutational approach effectively converted the enzyme to a form able to inhibit platelet aggregation, similar to the blood-feeding arthropod apyrases (Figure 4). Reversal of ADP-induced platelet aggregation was also observed using the mutant HB enzyme. Thus, the functional similarity of the vertebrate and invertebrate enzymes is clearly demonstrated through the mutation of just a few amino acids.

Inhibition and reversal of ADP-induced platelet aggregation using wild-type and HB (human/bedbug) mutant CAN. Platelet aggregometry and disaggregation studies were performed using human platelet-rich plasma treated with buffer (control), wild-type human CAN, or mutant G160S/L161S/K163M/T168K/E178M (denoted HB CAN). (A) In the presence of buffer (control, blue line, 0 µg/ml) platelets are strongly aggregated in response to ADP (10 µM final concentration added at 1 min, indicated by an arrowhead). HB CAN (blue traces) abrogates the ADP-induced platelet aggregation in a concentration-dependent manner, with complete blockade of aggregation at 20 µg/ml enzyme. However, wild-type human CAN (red trace) lacks the ability to prevent platelet aggregation even at a concentration of 400 µg/ml. (B) HB CAN was also tested for its ability to disrupt platelet aggregates. Addition of 10 µM ADP (arrowhead) to platelet-rich human plasma resulted in maximal platelet aggregation within the first 4 min. HB CAN was then added after induction of platelet aggregation by ADP at 3 min, and the resultant dissociation of the platelet aggregates was measured by the decrease in optical density of the sample. The addition of increasing amounts of the HB CAN after initiation of platelet aggregation resulted in increasing platelet disaggregation. Wild-type human CAN (red trace) lacks the ability to reverse platelet aggregation, even at a concentration of 400 µg/ml. Reprinted from Cell, Vol. 116, Dai et al., Structure and Protein Design of a Human Platelet Function Inhibitor, pp 649–659, Copyright (2004), with permission from Elsevier.
Therapeutic possibilities
The platelet is a major contributor to occlusive thrombus formation in acute coronary syndromes and stroke in humans, which are the primary causes of death and disability in the industrialized world. The major platelet inhibitors currently approved for use in thrombotic disorders by definition target the platelet itself, with a predominant side effect often being bleeding complications (reviewed in [22–24]). Achieving a potent anti-thrombotic effect without undermining hemostasis would be a ‘magic bullet’ against arterial thrombosis [24], thus new approaches for anti-platelet therapy utilizing soluble apyrases are being examined. One potential advantage of soluble nucleotidases that preferentially hydrolyze adenosine-based nucleotides is that they remove one of the major factors of the platelet release reaction (ADP), effectively limiting further platelet aggregation in a manner that does not involve binding to the platelet surface or disruption of intracellular protein machinery. Thus, serious bleeding complications may be minimized using this approach. It is important to point out however, that the vertebrate CAN proteins described in this review are primarily UDP/GDP hydrolyzing enzymes, with little endogenous ADPase activity (hence, their physiological function likely does not involve control of hemostasis). However, the bloodfeeding arthropod enzymes and also the HB mutant constructed by Dai et al. [20] are potent ADPases, capable of preventing ADP-mediated platelet aggregation.
There have been several recent reviews on the utilization of ADP hydrolyzing enzymes (usually a soluble derivative of the endothelial cell CD39/NTPDase1 apyrase, unrelated to the calcium activated nucleotidase (CAN) family members described in this report) in occlusive vascular disorders [25–30]. The therapeutic utility of these apyrases has been demonstrated in a variety of cardio- and cerebrovascular disorders, including middle cerebral artery occlusion [31, 32], endothelial denudation-induced arterial thrombosis [33], ATP and ischemia-induced norepinephrine release from heart synaptosomes [29], intestinal ischemia-reperfusion injury [34], arterial balloon injury [35], and cardiac xenograft transplantation [36, 37]. Hence, the unique anti-platelet actions of the ADP hydrolyzing enzymes make them particularly attractive as soluble protein therapeutics for the inhibition of platelet-mediated thrombotic pathologies.
Summary and conclusions
In this review, we have summarized the characterization of a distinct new family of nucleotidases with representative members found in both vertebrates and invertebrate bloodfeeding arthropods. Due to the strict dependency on calcium reported for all characterized family members to date, the designation CAN (calcium activated nucleotidase) has been proposed for this family. Enzymatic studies of the proteins isolated from different species have made it very clear however that the invertebrate and vertebrate members of this family do not share common nucleotide substrate preferences, presumably related to their distinct physiological functions. It is also interesting to note that the amino acid sequences of this novel family of enzymes have no homology to the NTPDase family of nucleotidases (e.g., CD39/NTPDase1) and their ‘apyrase conserved regions’ (ACRs [15]). Thus, the CANs are enzymatically analogous, rather than homologous in sequence, to the NTPDases, performing similar enzymatic functions despite their evolutionary distance. The differences in amino acid sequences between these two families of nucleotidases are also reflected in dramatic differences in their determined/predicted tertiary structures (see the article on NTPDase3 mutagenesis and structure by Kirley et al. appearing in this volume). Thus, we extend the observation made by Valenzuela et al. [5], that the CAN and the NTPDase families of nucleotide hydrolyzing enzymes are the product of convergent evolution, having evolved proteins of similar biochemical function from unrelated genes, which give rise to unrelated 3-D protein structures.
Abbreviations
- ADP:
-
adenosine diphosphate
- CAN:
-
calcium activated nucleotidase
- EST:
-
expressed sequence tag
- GDP:
-
guanosine diphosphate
- NCR:
-
nucleotidase conserved region
- NTPDase:
-
nucleoside triphosphate diphosphohydrolase
- SCAN:
-
soluble calcium activated nucleotidase
References
Cheeseman MT. Characterization of apyrase activity from the salivary glands of the cat flea Ctenocephalides felis. Insect Biochem. Mol. Biol. 1998; 28: 1025–0.
Mans BJ, Gaspar AR, Louw AI et al. Apyrase activity and platelet aggregation inhibitors in the tick Ornithodoros savignyi (Acari: Argasidae). Exp Appl Acarol 1998; 22: 353–56.
Perez de Leon AA, Tabachnick WJ. Apyrase activity and adenosine diphosphate induced platelet aggregation inhibition by the salivary gland proteins of Culicoides variipennis, the North American vector of bluetongue viruses. Vet Parasitol 1996; 61: 327–38.
Charlab R, Valenzuela JG, Rowton ED et al. Toward an understanding of the biochemical and pharmacological complexity of the saliva of a hematophagous sand fly Lutzomyia longipalpis. Proc Natl Acad Sci USA 1999; 96: 15155–60.
Valenzuela JG, Belkaid Y, Rowton E et al. The salivary apyrase of the blood-sucking sand fly Phlebotomus papatasi belongs to the novel Cimex family of apyrases. J Exp Biol 2001; 204: 229–37.
Basanova AV, Baskova IP, Zavalova LL. Vascular-platelet and plasma hemostasis regulators from bloodsucking animals. Biochemistry (Mosc) 2002; 67: 143–50.
Champagne DE. Antihemostatic strategies of blood-feeding arthropods. Curr Drug Targets Cardiovasc Haematol Disord 2004; 4: 375–96.
Ribeiro JM, Francischetti IM. Role of arthropod saliva in blood feeding: Sialome and post-sialome perspectives. Annu Rev Entomol 2003; 48: 73–88.
Valenzuela JG, Charlab R, Galperin MY et al. Purification, cloning, and expression of an apyrase from the bed bug Cimex lectularius. A new type of nucleotide-binding enzyme. J Biol Chem 1998; 273: 30583–90.
Kunapuli SP. P2 receptors and platelet activation. Sci World J 2002; 2: 424–33.
Murugappan S, Shankar H, Kunapuli SP. Platelet receptors for adenine nucleotides and thromboxane A2. Semin Thromb Hemost 2004; 30: 411–8.
Benoit P, Dogne JM. Platelet ADP receptors and their antagonists. Mini Rev Med Chem 2003; 3: 145–8.
Hechler B, Cattaneo M, Gachet C. The P2 receptors in platelet function. Semin Thromb Hemost 2005; 31: 150–61.
Failer BU, Braun N, Zimmermann H. Cloning, expression, and functional characterization of a Ca(2+)-dependent endoplasmic reticulum nucleoside diphosphatase. J Biol Chem 2002; 277: 36978–86.
Handa M, Guidotti G. Purification and cloning of a soluble ATP-diphosphohydrolase (apyrase) from potato tubers (Solanum tuberosum). Biochem Biophys Res Commun 1996; 218: 916–23.
Smith TM, Hicks-Berger CA, Kim S et al. Cloning, expression, and characterization of a soluble calcium-activated nucleotidase, a human enzyme belonging to a new family of extracellular nucleotidases. Arch Biochem Biophys 2002; 406: 105–15.
Murphy DM, Ivanenkov VV, Kirley TL. Bacterial expression and characterization of a novel, soluble, calcium-binding, and calcium-activated human nucleotidase. Biochemistry 2003; 42: 2412–21.
Yang M, Kirley TL. Site-directed mutagenesis of human soluble calcium-activated nucleotidase 1 (hSCAN-1): Identification of residues essential for enzyme activity and the Ca(2+)-induced conformational change. Biochemistry 2004; 43: 9185–94.
Murphy-Piedmonte DM, Crawford PA, Kirley TL. Bacterial expression, folding, purification and characterization of soluble NTPDase5 (CD39L4) ecto-nucleotidase. Biochim Biophys Acta 2005; 1747: 251–9.
Dai J, Liu J, Deng Y et al. Structure and protein design of a human platelet function inhibitor. Cell 2004; 116: 649–59.
Mans BJ, Coetzee J, Louw AI et al. Disaggregation of aggregated platelets by apyrase from the tick, Ornithodoros savignyi (Acari: Argasidae). Exp Appl Acarol 2000; 24: 271–82.
Ulrichts H, Vanhoorelbeke K, Van De Walle G et al. New approaches for antithrombotic antiplatelet therapies. Curr Med Chem 2004; 11: 2261–3.
Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov 2003; 2: 15–28.
Jackson SP, Schoenwaelder SM. Antiplatelet therapy: In search of the ‘magic bullet’ Nat Rev Drug Discov 2003; 2: 775–89.
Robson SC, Wu Y, Sun X et al. Ectonucleotidases of CD39 family modulate vascular inflammation and thrombosis in transplantation. Semin Thromb Hemost 2005; 31: 217–33.
Dwyer KM, Robson SC, Nandurkar HH et al. Thromboregulatory manifestations in human CD39 transgenic mice and the implications for thrombotic disease and transplantation. J Clin Invest 2004; 113: 1440–6.
Qawi I, Robson SC. New developments in anti-platelet therapies: Potential use of CD39/vascular ATP diphosphohydrolase in thrombotic disorders. Curr Drug Targets 2000; 1: 285–96.
Marcus AJ, Broekman MJ, Drosopoulos JH et al. Role of CD39 (NTPDase-1) in thromboregulation, cerebroprotection, and cardioprotection. Semin Thromb Hemost 2005; 31: 234–46.
Marcus AJ, Broekman MJ, Drosopoulos JH et al. Metabolic control of excessive extracellular nucleotide accumulation by CD39/ecto-nucleotidase-1: Implications for ischemic vascular diseases. J Pharmacol Exp Ther 2003; 305: 9–16.
Weksler BB. Antiplatelet agents in stroke prevention. Combination therapy: Present and future. Cerebrovasc Dis 2000; 10 Suppl 5: 41–8.
Pinsky DJ, Broekman MJ, Peschon JJ et al. Elucidation of the thromboregulatory role of CD39/ectoapyrase in the ischemic brain. J Clin Invest 2002; 109: 1031–40.
Belayev L, Khoutorova L, Deisher TA et al. Neuroprotective effect of SolCD39, a novel platelet aggregation inhibitor, on transient middle cerebral artery occlusion in rats. Stroke 2003; 34: 758–63.
Costa AF, Gamermann PW, Picon PX et al. Intravenous apyrase administration reduces arterial thrombosis in a rabbit model of endothelial denudation in vivo. Blood Coagul Fibrinolysis 2004; 15: 545–51.
Guckelberger O, Sun XF, Sevigny J et al. Beneficial effects of CD39/ecto-nucleoside triphosphate diphosphohydrolase-1 in murine intestinal ischemia-reperfusion injury. Thromb Haemost 2004; 91: 576–86.
Gangadharan SP, Imai M, Rhynhart KK et al. Targeting platelet aggregation: CD39 gene transfer augments nucleoside triphosphate diphosphohydrolase activity in injured rabbit arteries. Surgery 2001; 130: 296–303.
Imai M, Takigami K, Guckelberger O et al. Recombinant adenoviral mediated CD39 gene transfer prolongs cardiac xenograft survival. Transplantation 2000; 70: 864–70.
Imai M, Takigami K, Guckelberger O et al. CD39/vascular ATP diphosphohydrolase modulates xenograft survival. Transplant Proc 2000; 32: 969.
Acknowledgement
This work was partially supported by Wyeth Discovery Research (T.M.S.) and by NIH grants HL59915 and HL72882 to T.L.K.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Smith, T.M., Kirley, T.L. The calcium activated nucleotidases: A diverse family of soluble and membrane associated nucleotide hydrolyzing enzymes. Purinergic Signalling 2, 327–333 (2006). https://doi.org/10.1007/s11302-005-5300-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-005-5300-7