Article Text

Abstract
Background Cockayne syndrome (CS) is a rare, autosomal recessive multisystem disorder characterised by prenatal or postnatal growth failure, progressive neurological dysfunction, ocular and skeletal abnormalities and premature ageing. About half of the patients with symptoms diagnostic for CS show cutaneous photosensitivity and an abnormal cellular response to UV light due to mutations in either the ERCC8/CSA or ERCC6/CSB gene. Studies performed thus far have failed to delineate clear genotype-phenotype relationships. We have carried out a four-centre clinical, molecular and cellular analysis of 124 patients with CS.
Methods and results We assigned 39 patients to the ERCC8/CSA and 85 to the ERCC6/CSB genes. Most of the genetic variants were truncations. The missense variants were distributed non-randomly with concentrations in relatively short regions of the respective proteins. Our analyses revealed several hotspots and founder mutations in ERCC6/CSB. Although no unequivocal genotype-phenotype relationships could be made, patients were more likely to have severe clinical features if the mutation was downstream of the PiggyBac insertion in intron 5 of ERCC6/CSB than if it was upstream. Also a higher proportion of severely affected patients was found with mutations in ERCC6/CSB than in ERCC8/CSA.
Conclusion By identifying >70 novel homozygous or compound heterozygous genetic variants in 124 patients with CS with different disease severity and ethnic backgrounds, we considerably broaden the CSA and CSB mutation spectrum responsible for CS. Besides providing information relevant for diagnosis of and genetic counselling for this devastating disorder, this study improves the definition of the puzzling genotype-phenotype relationships in patients with CS.
- ercc6
- ercc8
- csa
- csb
- hotspot
Statistics from Altmetric.com
Introduction
Cockayne syndrome (CS) (OMIM #216400 and #133540) is a rare autosomal recessive disorder characterised by severe developmental delay, mental retardation, microcephaly, cachexia and a variety of other features, which may include cataracts, retinal degeneration, sensorineural hearing loss, dental anomalies and photosensitivity.1–5 There is a large variation in severity of the disorder, which has led to categorisation into three types: type I is associated with normal features at birth, followed by the onset of clinical features starting in the first or second year of life. The clinical features are progressive, usually leading to death in the second or third decade of life. Type II represents a more severe form of the disorder with features present at birth or prenatally. This group typically does not survive beyond the first decade. Type III represents a group with less severe features than those in type I. They may survive for several decades. Cerebro-oculo-facio-skeletal syndrome has also been used to describe a very severe form of the disorder. This categorisation is quite convenient for a rough description of the patients’ severity,2 but in reality there is probably a continuum of severity of features.6
At the cellular level, a robust diagnostic test is provided by the response of RNA synthesis to ultraviolet (UV) irradiation of cultured fibroblasts.7 Whereas RNA synthesis and subsequently DNA synthesis recover rapidly following UV irradiation of normal fibroblasts, this recovery is much delayed or absent in CS fibroblasts.8 Nearly all cases in which there is a clear clinical diagnosis of CS are defective in this test, and in almost all patients diagnosed by this test, the causative mutation lies in one of two genes, ERCC6/CSB (OMIM 609413) or ERCC8/CSA (OMIM 609412). The encoded proteins, CSB and CSA are respectively a DNA-dependent ATPase9 10 and a WD40 protein component of a large cullin4-mediated E3-ubiquitin ligase complex.11 12 The ATPase activity of the 1493 aa CSB protein falls into the SWI2/SNF family and is associated with seven so-called helicase domains, even though CSB does not have helicase activity. Towards the C-terminus there is a ubiquitin-binding domain.13 CSB can be modified by phosphorylation, ubiquitylation on lys99114 and SUMOylation, most likely on lys205.15 16 The 396 aa CSA protein comprises a seven-bladed WD40 propeller attached to the DDB1 protein via a helix-loop-helix motif at the N-terminus.12
The best characterised role of the CS proteins is in the transcription-coupled branch of nucleotide excision repair (NER) of UV-induced DNA damage. This damage, when in the transcribed strand of active genes, results in stalling of RNA polymerase II. The CS proteins are thought to modify the chromatin in the region of the stalled polymerase, enabling the polymerase to back-track and then to assist in the recruitment of TFIIH and other proteins involved in subsequent steps of NER.17 18 This role of the CS proteins readily explains the failure of RNA synthesis to recover following UV irradiation of CS cells and the photosensitivity of the patients. However, it is not so easy to reconcile with many of the other features of CS. Indeed a few patients, with so-called UV-sensitive syndrome (UVSS), have been identified with mutations in CSA, CSB or a recently identified gene UVSSA.19–23 Cells from these individuals show the same defective recovery of RNA synthesis as CS cells but the patients display only the sun-sensitivity and not the broad spectrum of other features of CS such as neurodegeneration and premature ageing.24 These observations suggest that the CS proteins have other functions as well, and evidence has been provided for several other roles,5 25 including the repair of oxidative damage in DNA26–29 and roles in mitochondrial DNA metabolism.30–33 A recent elegant study, using both whole brains and cultured cells, identified a crucial role for the CS proteins in expression of neuronal genes and thereby in neuronal differentiation.34 Similar conclusions have been reached from a study in which induced pluripotent stem cell-derived neuronal cells from patients with CS had reduced transcription of many neural-specific genes.35 This role of CS proteins in neuronal differentiation could account for some of the developmental defects found in patients with CS.
In this manuscript, we have gained further insight into the genetics and molecular basis of CS by analysing the clinical features and mutations in 124 patients with CS, combining data gathered over several decades from four centres, in Strasbourg (France); Pavia (Italy); Nagasaki and Nagoya (Japan) and Brighton (UK). Our results have identified many novel genetic variants and provide insights into previously unreported genotype-phenotype relationships and their relevance for clinical diagnosis.
Materials and methods
Samples were obtained as skin biopsies, fibroblast cultures, blood or DNA extracted from blood, all with appropriate informed consent.
Fibroblast cultures and lymphoblastoid cell lines were established from skin biopsies and blood lymphocytes, respectively, and grown using standard procedures. In the Pavia, Nagasaki and Brighton labs, cells were first screened on a diagnostic basis using the post-UV recovery of RNA synthesis (RRS) test using liquid scintillation counting,7 36 autoradiography37 or a fluorescence assay.7 36 Several cell samples were also analysed for hypersensitivity to the killing effects of UV exposure and levels of UV-induced DNA repair synthesis (UDS). Only cells displaying a defective RRS were characterised further to identify the mutation in the defective CS gene. In Pavia, cell fusion with known CS-A or CS-B cells using polyethylene glycol was used to establish complementation group.37 In Japan, complementation group was established by transduction with lentivirus expressing either ERCC6/CSB or ERCC8/CSA cDNA.38 Finally, the appropriate gene was sequenced using genomic DNA (ERCC6 RefSeq NG_009442.1; ERCC8 RefSeq NG_009289.1) and/or cDNA (ERCC6 NM_000124.3; ERCC8 RefSeq NM_000082.3). In Strasbourg, RRS and molecular screening (genomic and/or cDNA sequence) were performed concomitantly. Genomic sequencing was performed either by Sanger or next-generation sequencing.39 Mutation nomenclature follows the format indicated at http://varnomen.hgvs.org/. Nucleotide numbering of coding sequences starts with the A of the ATG translation initiation site as nucleotide 1. When appropriate, we consulted the Human Splicing Finder (HSF), a tool to predict the effects of mutations on splicing signals or to identify splicing motifs in any human sequence (http://www.umd.be/HSF3/HSF.shtml).
Clinical examination was carried out by VL and colleagues for all patients analysed in Strasbourg. Descriptions of clinical features at the other centres were dependent on clinical notes supplied by the referring clinicians.
Results and discussion
Out of the 124 patients identified as having a specific defect in RRS, 39 were mutated in ERCC8/CSA (table 1) and 85 in ERCC6/CSB (table 2), representing 32% and 68% of the population, respectively. Homozygous patients (30 CS-A and 43 CS-B) are listed first in order of mutation position. Compound heterozygotes (9 CS-A and 42 CS-B) are listed subsequently in order of the most 5’ of the two genetic variants. Tables 1 and 2 also summarise as much clinical data as we have available, including previous reports on 13 CS-A and 5 CS-B cases.
Mutation and clinical data on patients with CS-A
Mutation and clinical data on patients witlh CS-B
CSA mutations
We have identified 32 pathogenic genetic variants in CSA, of which 25 have not been reported previously. Six were missense mutations, all but one previously unreported, and one a small in-frame indel. Missense mutations are indicated below the CSA linear structure (figure 1A) with the previously unreported alterations indicated in bold. CSA comprises a seven-bladed WD40 propeller attached to the DDB1 protein via a helix-loop-helix motif at the N-terminus.12 All the newly identified missense mutations affect residues that are conserved among CSA orthologs and are located in WD40 repeats. Together with previously reported mutations, indicated above the CSA linear structure, there is a particularly high concentration of missense mutations around aa 200 (4 mutations within 12 aa) and aa 270 (3 mutations within 7 aa). All the 13 missense mutations are located in the blades of the beta propeller structure, with 8 of them clustered in blades 4 and 5 (figure 1B), and are likely to disrupt the structure of the protein.12

Mutation distribution in CSA. (A) Distribution of missense mutations across the CSA protein. Mutations identified in this study are indicated below the protein with new mutations indicated in bold. Other previously identified missense mutations are indicated above the protein; 1–7 indicate the seven WD40 domains of the CSA protein according to the reference sequence NP_000073.1. (B) Missense mutations associated with different CS phenotypes mapped onto the three-dimensional CSA protein structure (RCSB PDB, DOI: 10.2210/pdb4a11/pdb11 12). Yellow, UVSS; orange, CS type I; violet, CS type II; dark grey, CS type III. (C) Distribution of truncation mutations. Mutations have been grouped in intervals of 40 aa and columns represent the number of mutations for each group. The interval 0–40 includes mutations resulting in no transcript (asterisks). Black: new mutations identified in this study; grey: previously reported mutations also present in this study; white: other previously reported mutations.
All the missense mutations are predicted to be pathogenic using Polyphen, MutPred2 and SIFT (see online supplemen tary table 1). We have confirmed their defective function by transducing a CS-A cell line, CS9LO, with virus containing the mutant cDNA (figure 2A, B). Wild-type CSA almost completely restored RRS to the recipient UV-irradiated CS9LO cell line. In contrast, when the cells were transduced with any of the six mutant cDNAs, RRS remained close to the level of the untransduced cells (figure 2A). The infection efficiency was similar for all transductions (figure 2B).
Supplementary file 1
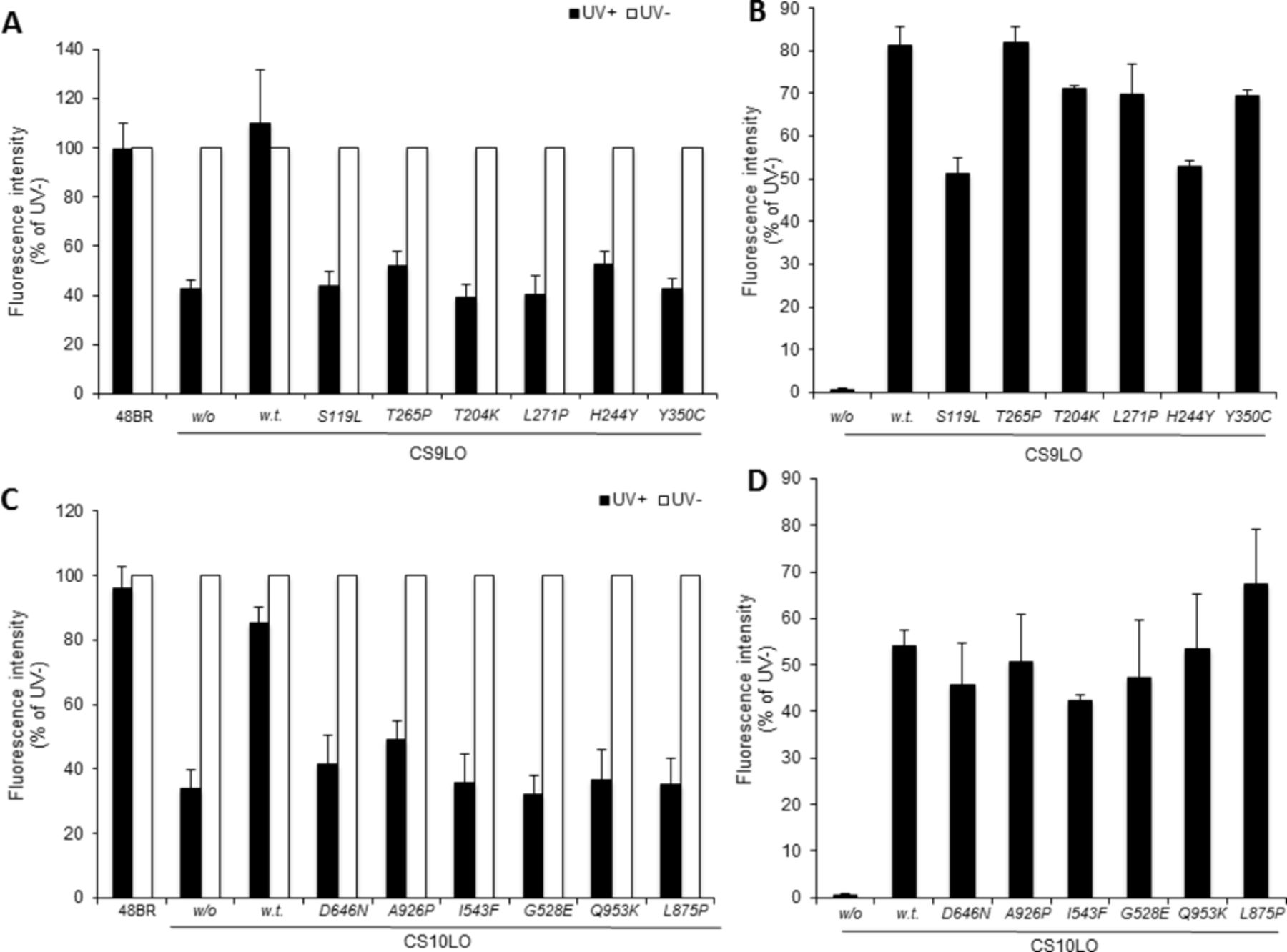
Lack of complementation with CSA and CSB mutations. Wild-type and various mutant ERCC8/CSA (A, B) or ERCC6/CSB (C, D) cDNAs were ectopically expressed by recombinant lentivirus infection in fibroblasts derived from a patient with CS-A, CS9LO or patient with CS-B, CS10LO, respectively. (A, C) Recovery of RNA synthesis activities were detected 12 hours after UV irradiation (filled bars, 12 J/m2 UVC irradiation; open bars, no UV irradiation), and the value was normalised to activity measurement in non-irradiated cells. (B, D) Viral infection efficiency was confirmed by immunofluorescent staining of V5-tagged wild-type and mutant CSA or CSB proteins, and calculated as the number of Alexa 488-positive cells using a semi-automatic VTI system. w/o, without virus infection; w.t., wild type. Results from at least three independent experiments. Error bars indicate SD.
We have identified 19 protein-truncating genetic variants, of which 14 are new, that include frameshift, splicing and premature stop mutations. They are predicted to result in 18 distinct truncated proteins (12 new), because we found that the Thr134Leufs*13 truncation (due to exon 5 deletion, ie, r.400_481del) is caused by two distinct mutations affecting either the splice acceptor site of intron 4 (c.400-2A>G in CS261ST) or the splice donor site of intron 5 (c.479C>T in CS133NY). In addition, two mutations resulting in large in-frame deletions (p.Val282_Gln347del, Val282_Glu374del and p.Val27_Arg92del) are respectively predicted from c.966C>A and a previously unreported rearrangement involving part of intron 2 and exon 3, which results in a transcript lacking exons 2 and 3 (patient CS1LE). Interestingly, the genomic mutation c.966C>A (in exon 10), previously described as resulting in a single, full-length normal-spliced transcript (r.966c>a, p.Tyr322*),39 was shown to generate also two abnormally spliced transcripts carrying the deletion of exons 10 and 11 (r.844_1122del; p.Val282_Glu374del in CS9IAF) and/or the deletion of exon 10 (r.844_1041del; p.Val282_Glu347del in CS5IAF and CS9IAF). Indeed, this mutation is predicted to alter an exonic splicing enhancer (ESE) site, and potentially alter the splicing as indicated by the bioinformatics tool HSF.
Finally, at the genomic level we identified three new CSA genetic variants located in splice donor sites (c.481+1G>C, c.1041+1G>T and c.1122+1 delG) most probably affecting splicing. The resulting transcripts could not be identified because of the unavailability of RNA samples. However, HSF analysis indicated potential splicing alterations for the three mutations as well as the activation of an intronic cryptic donor site for the latest two.
CSB mutations
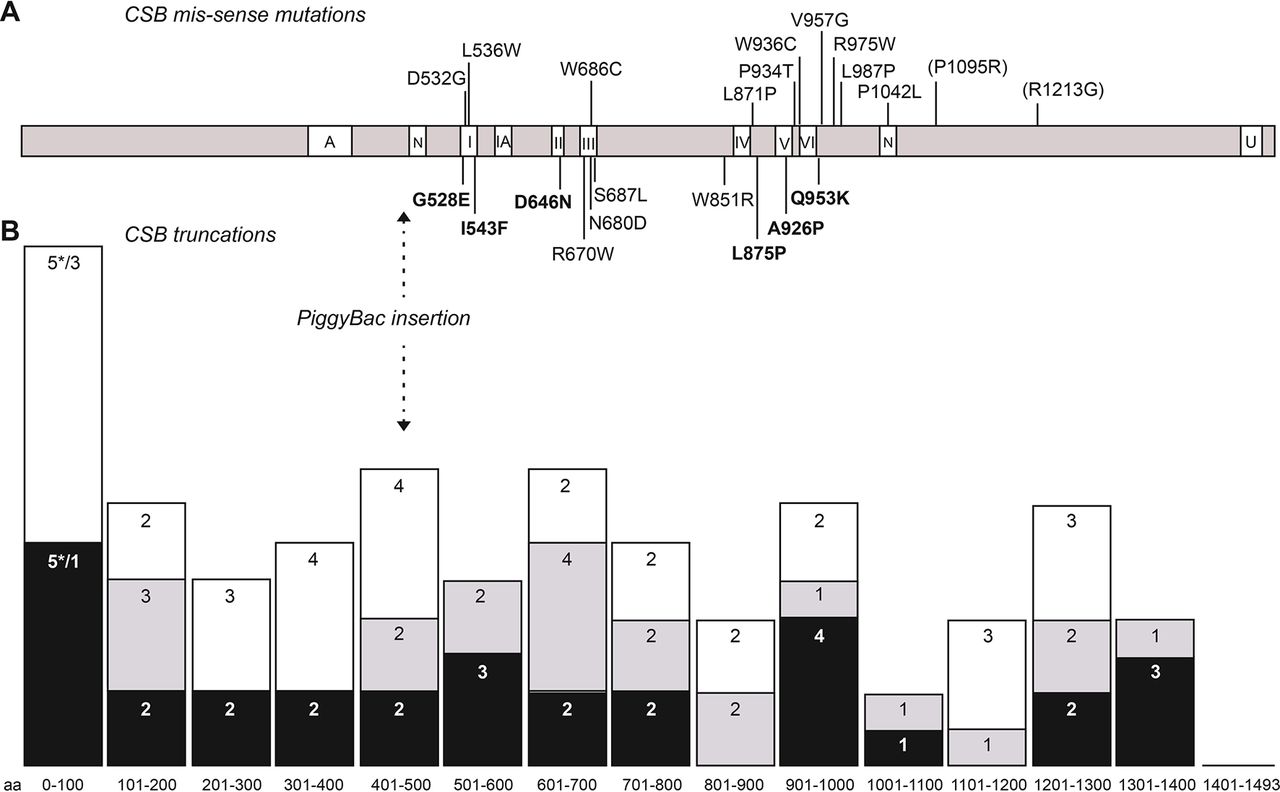
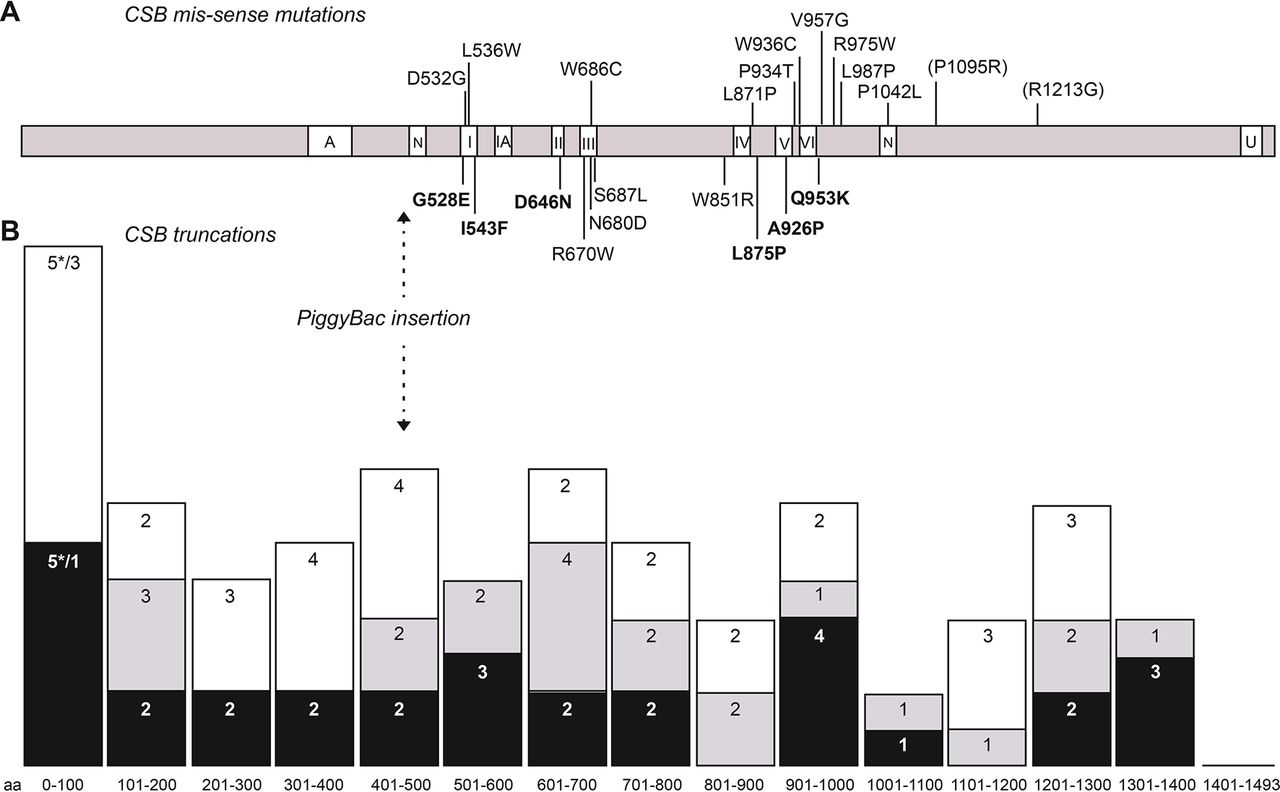
In CSB, we identified 73 pathogenic genetic variants of which 46 were previously unreported. Ten of the mutations were missense, 9 in-frame deletions (2 small and 7 large), 5 null mutations leading to unexpressed transcripts and the rest being truncations resulting from stop (16), frameshifts (20) or splice mutations (13). The 10 missense mutations are indicated in figure 3A, below the CSB linear structure, with the 6 new mutations indicated in bold. Other previously reported missense mutations are shown above the CSB linear structure.
{kind=link}
{kind=link}
{kind=link}
Mutation distribution in CSB. (A) Distribution of missense mutations across the CSB protein. Mutations identified in this study are indicated below the protein with new mutations indicated in bold. Other missense mutations reported as pathogenic are indicated above the protein, with those now classified as polymorphic variants in parenthesis. Different domains of the protein are indicated: A, acidic domain; N, nuclear localisation domain; I, IA, II–VI, helicase-like domains; U, ubiquitin-binding domain. (B) Distribution of truncation mutations. Mutations have been grouped in intervals of 100 aa and columns represent the number of mutations number for each group. The interval 0–100 includes mutations resulting in no transcript (asterisks). Black: new mutations identified in this study; grey: previously reported mutations also present in this study; white: other previously reported mutations.
All missense mutations are predicted to be pathogenic (see online supplementary table 1). We have confirmed the defective function of several of them by transducing a CS-B cell line, CS10LO, with virus containing the mutant cDNA (figure 2C, D). Similar to the CSA data, wild-type CSB almost completely restored RRS to the recipient UV-irradiated CS10LO cell line, whereas with the six mutant cDNAs tested, RRS remained close to the level of the untransduced cells (figure 2C). The infection efficiency was similar for all transductions (figure 2D).
The distribution of CSB truncation mutations is presented in figure 3B (lower panel). Some general conclusions may be drawn from the present in-depth investigation together with previous studies (data up to 2010 reviewed by Laugel et al,39 and since 2011 listed in online supplementary table 2). With two exceptions, all of the CSB missense mutations are located either in or very close to the seven helicase domains, in particular in domains I and III (four mutations each) and domains IV–VI (9 mutations within 110 amino acids), emphasising the crucial role of these domains in CSB function. These helicase domains are involved in the DNA-dependent ATPase activity of the protein and confirm that this activity is vital for preventing the features of CS. The N-terminal and C-terminal extensions are likely to be much more amenable to genetic variants that do not affect function, despite the demonstration that the C-terminal part of the protein is essential for a normal cellular response to UV irradiation.13 16 This may imply that the structure of the C-terminal ubiquitin—binding domain needs to be intact, but the precise amino acid sequence is less crucial. Again, as might be anticipated, the truncation mutations are spread rather evenly across the protein (figure 3B).
Supplementary file 2
Interestingly, the c.1834C>T and c.2143G>T result in p.Arg612* and p.Gly715*, respectively, and their transcript was detected only in homozygous or hemizygous patients. This transcript must be poorly expressed, presumably because of nonsense-mediated decay, because in compound heterozygotes only the transcript resulting from the second allele was detected.
Furthermore, several genetic variants affect ERCC6/CSB splicing giving rise to either truncations or in-frame deletions. In particular, 10 mutations map at the canonical splice sites of different exons, four are located inside introns (c.1993-5A>G; c.2599-26A>G, c.1685+6T>G, c.543+4 delA) and three inside exons (c.466C>T, c.526C>T and c.2092_2093insG). All the exonic changes are likely to alter the splicing by creating novel exon-splicing enhancer (ESE in the case of c.466C>T) or exon-splicing silencer (ESS for c.526C>T and c.2092_2093insG) sites according to HSF prediction.
Recurring pathogenic genetic variants
Mutations found in several patients are indicative of either founder effects or mutation hotspots. Although haplotype analysis would be required to distinguish definitively between these two alternatives, as a first approximation, we assume that if the pathogenic genetic variant is only found in a relatively limited geographical location, it is more likely to be a founder effect. In ERCC8/CSA, we found a complex rearrangement involving exon 4 in 8 Japanese patients, also previously reported in four Japanese patients by Ren et al,40 strongly indicating a founder mutation. Moreover, the c.966C>A mutation found in two patients of our cohort (CS9IAF, CS5IAF) was also previously described in three cases (CS2IAF, CS886VI/CS887VI).39 Since all the patients are of Arabic origin, although from different countries (Israel or Lebanon), this is also most likely a founder mutation.
Nine CSB/ERCC6 pathogenic genetic variants occur in three or more patients (table 3). The most common of these multiple occurrences are c.2203C>T, c.2167C>T and c.466C>T, respectively found in 12, 11 and 7 patients. Whereas c.2167C>T and c.466C>T are found almost exclusively in the UK patients and may result from founder effects, c.2203C>T is found in individuals from several different countries and likely results from independent mutations. Interestingly, the C>T mutations in table 3 that are more likely to result from a founder mutation are at CpA sites, whereas those more likely to result from independent mutations are at CpG sites. CpG sites are known to be mutational hotspots in the human genome.41
ERCC6/CSB mutations identified in three or more patients
Relationship to clinical features
No obvious genotype-phenotype correlation was identified in the patients with CS-A reported in previous investigations (45 cases from 33 families). With the present study, we have expanded the cohort of patients with CS-A by describing 39 new cases, the majority of which are homozygotes. Focusing on the homozygous patients with CS-A (33 from 24 families in the literature and 30 from 30 families in our cohort, excluding the 9 Japanese cases with a recurrent mutation), missense mutations appear to be more frequently associated with mild phenotypes than protein-truncating mutations. The observation that the missense alteration p.Trp361Cys, which interferes with transcription-coupled NER but not with the oxidative stress response, is associated with UVSS, a rare disorder characterised only by cutaneous photosensitivity,20 strongly supports the notion that the severity of the clinical features is related to the effects of the mutation on the additional roles of CSA outside transcription-coupled NER, which include oxidative damage response, mitochondrial function maintenance and ribosomal DNA transcription.
Previous analysis of mutations in patients with CS-B (51 homozygous cases from 29 families/kindreds and 37 compound heterozygotes from 32 families) have not identified any clear correlation between the site or the nature of the mutations with the type and severity of the clinical features,42 43 although some more subtle relationships have been suggested. Several years ago, Horibata et al suggested that CSB truncations generating no functional protein resulted in the mild phenotype of UVSS, whereas more C-terminal truncations might generate inactive protein that could interfere with other processes, thereby resulting in more severe phenotypes.19 Weiner and colleagues showed that the human ERCC6/CSB gene contains a PiggyBac transposon insertion in intron 544 45 (see table 2 and figure 3). They showed that translation of ERCC6/CSB resulted in bona fide CSB protein, but also a CSB-PiggyBac fusion protein. Truncation mutations upstream of intron 5 would generate neither protein, whereas those downstream would generate only the CSB-PiggyBac fusion, which was proposed to have deleterious effects.44 We have analysed the severity of the clinical features in our patient cohort to see if they are in accord with these suggestions. In eight patients homozygous for truncations in the first five exons, six could be categorised as type I, and one as type II. No information is available for one patient. In contrast, in 28 patients homozygous for truncations downstream of exon 5, the numbers assigned to types I, II and III are 5, 12 and 3, respectively. There thus appears to be a tendency to more severe phenotypes (type II) associated with downstream truncations, although this does not seem to be an absolute correlation. Patients with truncations upstream of the Piggy-Bac insertion but severe clinical features have been reported previously.46 Furthermore, of the four patients homozygous for the mutation Asp1355Valfs*32, two were classified as type I and two as type III (see table 2). Altogether, these observations indicate that other factors, apart from the site of mutation, contribute to the severity of the pathological phenotype.
In an earlier analysis, Laugel suggested that type II features were more prevalent in patients with CS-B than in patients with CS-A.6 This is supported by our current data. The distributions for those patients for whom we have clinical data for types I, II and III are 67%, 21% and 12.5% for CS-A (21 patients) and 35%, 56% and 10% for CS-B (60 patients), respectively. The individual clinical features for which we have information are summarised in table 4, where they are also compared, where possible, with data from a recent analysis of 102 CS individuals by Wilson et al.4 Within our own cohort, there are few differences between patients with CS-A and CS-B, with the possible exceptions of cataracts, low birth weight and microphthalmia, which are more prevalent in patients with CS-B. The incidence of several features appears to be higher in our cohort than in that studied by Wilson et al (see table 4). Two possible explanations for this are: (1) they could represent genuine differences between the two cohorts; (2) the analytical clinical criteria may differ between the two studies. Of the patients subjected to molecular analysis in4 the ratio of CS-B to CS-A cases is very similar to that reported here.
Summary of clinical features
In a recent survey of patients with CS in Japan, nearly all of them (41/47) were categorised clinically as type I.3 Unfortunately, this survey did not include molecular analyses. However, our data strongly suggest that there is a ERCC8/CSA founder mutation in Japanese patients with CS. We may extrapolate this to suggest that many of the patients analysed in the survey by Kubota et al are likely also to have carried this founder mutation. As mentioned above, patients with CS-A are more likely to fall into the type I category. The features of the 41 Japanese patients with type I CS are also included in table 4. Deafness, photosensitivity and retinal degeneration appear to be higher in the Japanese cohort. This may be partially explained by the average age of the Japanese patients (17.5 years), which appears to be significantly higher than in our cohort. Deafness and retinal degeneration are progressive and therefore more likely to occur in older patients.
As also reported in earlier studies, clinical photosensitivity was found in the majority of our patients, even those with skin types IV and V on the Fitzpatrick Skin Type Scale (see tables 1, 2 and 4). Nevertheless, as in other reports,47 we found no skin cancers in any of our patients. This may be explained by a recent finding that CS fibroblasts are not hypermutable by UV radiation.48
In conclusion, our analyses show that the human mutation spectrum of the CS genes is not yet saturated, but missense mutations are largely confined to a few relatively short regions. There are no definitive correlations between genotype and phenotype, but truncation mutations C-terminal to the PiggyBac insertion in ERCC6/CSB are more likely to confer a severe clinical phenotype than mutations N-terminal to this insertion or mutations in ERCC8/CSA.
Acknowledgments
The authors are grateful to all the patients and referring clinicians for the samples used in this study, and to Roberta Ricotti for technical support.
References
Footnotes
NC, EB and NJ contributed equally.
Contributors NC, EB, NJ and CO carried out the sequence analysis; HF, TN, ML and CO did the RRS measurements and contributed to the genetic analysis, as did YN and NJ, who also did the lentivirus complementation. SM, KS, MK, MAS, MS, VL and ARL provided patient material and clinical data; MS, VL, DO, TO and ARL conceived and designed the study, and, together with NC and EB, wrote the paper. All authors approved the final manuscript.
Funding This work was funded by Associazione Italiana per la Ricerca sul Cancro Grant IG 13537 (MS) and IG 17710 (DO); Collaborative Projects on Rare Diseases by Istituto Superiore Sanità 526D/17 IST-CNR (MS); a grant for Research for overcoming intractable diseases (H26-general-046) from The Ministry of Health Labour and Welfare of Japan, KAKENHI Grants-in-Aid for Scientific Research (B) (26291005) from JSPS, KAKENHI Grants-in-Aid for Scientific Research (A) (Overseas Academic Research) (15H02654) from JSPS, a science research grant from the Uehara Memorial Foundation, and a scientific research grant from Daiko Foundation to TO and a grant from Agence de la Biomedecine to VL and NC.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Work from the UK and Japan had approval from the Ethics Research Committees of the University of Sussex and the Ethics Committee for Human Genome Studies in the Research Institute of Environmental Medicine, Nagoya University. The results from Strasbourg and Pavia are from retrospective collections of clinical and molecular data, which do not require specific ethics committee approval at these institutions for this particular work. All patient clinical data have been obtained in a manner conforming with IRB and granting agency ethical guidelines.
Provenance and peer review Not commissioned; externally peer reviewed.