Article Text

Abstract
Background Oral clefts, that is, clefts of the lip and/or cleft palate (CL/P), are the most common craniofacial birth defects with an approximate incidence of ~1/700. To date, physicians stratify patients with oral clefts into either syndromic CL/P (syCL/P) or non-syndromic CL/P (nsCL/P) depending on whether the CL/P is associated with another anomaly or not. In general, patients with syCL/P follow Mendelian inheritance, while those with nsCL/P have a complex aetiology and, as such, do not adhere to Mendelian inheritance. Genome-wide association studies have identified approximately 30 risk loci for nsCL/P, which could explain a small fraction of heritability.
Methods To identify variants causing nsCL/P, we conducted whole exome sequencing on 84 individuals with nsCL/P, drawn from multiplex families (n=46).
Results We identified rare damaging variants in four genes known to be mutated in syCL/P: TP63 (one family), TBX1 (one family), LRP6 (one family) and GRHL3 (two families), and clinical reassessment confirmed the isolated nature of their CL/P.
Conclusion These data demonstrate that patients with CL/P without cardinal signs of a syndrome may still carry a mutation in a gene linked to syCL/P. Rare coding and non-coding variants in syCL/P genes could in part explain the controversial question of ‘missing heritability’ for nsCL/P. Therefore, gene panels designed for diagnostic testing of syCL/P should be used for patients with nsCL/P, especially when there is at least third-degree family history. This would allow a more precise management, follow-up and genetic counselling. Moreover, stratified cohorts would allow hunting for genetic modifiers.
- expanded spectrum of syCL/P
- gene
- cleft
- syndrome
- mutation
- phenotype
- nsCL/P
- syCL/P
- WES
- NGS
- transcription factor
- craniofacial syndrome
- Van der Woude
- EEC
- 22qdel
- tooth agenesis (oligodontia)
Statistics from Altmetric.com
- expanded spectrum of syCL/P
- gene
- cleft
- syndrome
- mutation
- phenotype
- nsCL/P
- syCL/P
- WES
- NGS
- transcription factor
- craniofacial syndrome
- Van der Woude
- EEC
- 22qdel
- tooth agenesis (oligodontia)
Introduction
Cleft lip and/or cleft palate (CL/P) are the most prevalent craniofacial birth defects. They have an approximate incidence of 1/700 live births, varying with ethnicity, gender and cleft type.1 Although CL/P is no longer associated with mortality in higher income countries,2 it is a debilitating condition requiring expensive, invasive and often treatment until the adult age.
Syndromic CL/P (syCL/P) is typically caused by Mendelian mutation with a strongly deleterious effect on the function of a single protein, such as IRF6,3 or by a deletion of a cluster of contiguous genes, such as del22q.11 (22qdel).1 According to OMIM, there are 148 syndromes associated with CL/P.
Non-syndromic CL/P (nsCL/P), in contrast, is thought to have a complex aetiology, with multiple predisposing genetic variants acting in concert with intrauterine environmental effects. Epidemiological data suggest that if a child is born with a CL/P, the risk of re-occurrence in future siblings is calculated to be about 4%. The risk of recurrence is similar if one parent has CL/P, 17% if one parent and one child are affected and 9% when two children are affected.4 Non-syndromic oral clefts constitute 70% of all CL/P subjects, of which 80% are sporadic and 20% are multiplex (familial) cases.1
A common hypothesis has been that a complex disease, such as nsCL/P, is due to accumulation of predisposing common variants with a minor allele frequency >5% in the affected subjects.5 Association studies have been performed to identify them, either focusing on candidate genes or loci disrupted by karyotype aberrations in rare patients.6 7 Genome-wide association studies (GWASs) have also been performed, and roughly 30 loci have been identified.8–18 However, all these loci together explain only a small percentage of nsCL/P’s heritability.19 Thus, the enigma of ‘missing heritability’ remains and animates an intense debate.
We hypothesised that part of the missing heritability could be explained by rare (private) variants that are not assayed by GWASs. Such variants can be numerous in the population, with a medium-to-high penetrance. Moreover, there is high variability in expressivity in most single-gene oral cleft syndromes suggesting influence of genetic modifiers. Thus, rare variants in genes causing syCL/P could explain part of nsCL/P. We went out to look for such variants in a cohort of 84 individuals affected with nsCL/P, drawn from multiplex families (n=46).
Methods
Patient selection
In close collaboration over 25 years with the multidisciplinary team at the Centre Labio-Palatin (Cliniques universitaires Saint-Luc), and a network of specialists practising in craniofacial centres worldwide, we have assembled a bio-bank of over 1300 samples of blood DNA from index subjects with different syndromic and non-syndromic CL/P. We have implemented systematic guidelines to collect samples uniformly. This included a detailed ascertainment of the clinical phenotype and family history by physicians, facilitated by a standardised questionnaire.
The database contains 275 families with CL/P, of which 43 families (16%) have an IRF6 mutation, all diagnosed with a Van der Woude syndrome (VWS), either a priori (when lip pits were evident) or a posteriori (when an IRF6 mutation was identified and minor atypical lower lip signs were identified in at least one family member following scrutinised clinical examination).20 The remaining 234 families that were negative for IRF6 mutation were candidates for whole exome sequencing (WES), in order to identify new causal gene(s). We selected a total of 84 individuals drawn from 46 families classified clinically as nsCL/P cases, for which we had enough high-quality DNA for WES. When possible, we chose two to four most distant relatives per family (26/46 families, 64 individuals). For 20 families, only the index patient was sequenced. The subphenotypes of these patients ranged from full-blown complete bilateral clefts of the lip and palate (BCLP) to a much more subtle velopharyngeal insufficiency (VPI). We had a total of 7 bilateral CLP, 16 unilateral CL/P, 8 unilateral cleft lip (CL), 33 cleft palate (CP), 7 CP-posterior, 2 submucous cleft palate (SMCP), 2 SMCP with bifid uvula and 9 VPI. All selected patients had standard karyotype and many had also been tested for 22qdel, and these tests were normal. All participants and their legal guardians (when necessary) gave informed consent for the participation in the study. This study was approved by La Commission d’Ethique Biomédicale Hospitalo-Facultaire, reference 2015/02NOV/572, of the medical faculty at University of Louvain, Brussels, Belgium.
Whole exome sequencing
Blood was drawn in EDTA tubes, and DNA was extracted from blood samples using Wizard Genomic DNA Purification Kit (Promega). WES was carried out by using 1 µg of genomic DNA per sample. Genomic DNA was fragmented into a library of small segments that can be uniformly and accurately sequenced in millions of parallel reactions. Exomes were captured using a commercial enrichment kit (Agilent SureSelectXT Human All Exon KitV5). Uncaptured DNA was washed off. Bounded DNA was clonally amplified and sequenced on Illumina HiSeq2000 to generate paired-end, 100 bp reads. Real-time image analysis and base calling were performed by sequence control software real-time analysis and CASAVA software V.1.8 (Illumina). We achieved a mean coverage of 61× without duplicates, with 81% of target covered at least 10×.
Data analysis
Generated reads were aligned to the human genome reference sequence (assembly GRCh37/hg19) by Burrows-Wheeler Aligner (see URLs). Picard Mark duplicates tool was used to flag duplicated reads (reads with a same start and end coordinates), which were subsequently removed from downstream analysis. SAM tools consolidated all BAM files (aligned reads). Local realignments around indels and base quality score recalibration were done with the Genome Analysis Toolkit (GATK) V.2.8. Variants were called using the GATK Unified Genotyper. Each called variant was annotated using Highlander software, an in-house bioinformatic framework (see URLs) (Helaers et al, under revision). On average, each patient had ±20 000–25 000 variants.
In silico analysis of called variants
We analysed variants in each family and in each sporadic subject independently. We retained variants for further analysis if they met the following criteria: (1) ≤1% in Exome Aggregation Consortium (ExAC), (2) ≤1% in 1000 genomes project, (3) ≤0.5% in Go-NL (genome of the Netherlands), (4) ≤1% in in-house controls (subjects affected with different condition than CL/P) and (5) estimated by visual inspection on Integrative Genomics Viewer/GATK scores as true variants.
As ‘likely pathogenic’ variants, we considered those with a high impact (canonical splice-site variants and those generating a premature termination codon, PTC (out-of-frame indels and nonsense variants)), or a moderate impact (missense), as predicted by SnpEff software. We prioritised variants in genes intolerant to loss-of-function (LoF) and/or variation, as estimated from the PLi values and z-scores derived from ExAC. We considered missense variants to have a deleterious effect when at least three out of six software (Sift, Polyphen, LRT, Mutation taster, Mutation assessor, FATHMM) predicted them as damaging. To maximise the conclusion that any novel, damaging variant would be causal, we focused on a list of about 500 (functional) candidate genes that we gathered from Jugessur and coworkers (ie, 357 biologically plausible, autosomal candidate genes for oral clefts),21 augmented by novel genes presented at recent craniofacial conferences and or publications. We used the Genome Aggregation Database (gnomAD) to evaluate the frequency of the identified variants in a larger control population.
For all variants that satisfied our criteria, we performed co-segregation analysis on all the affected and non-affected family members when available. Approximately 15–20 variants/family and ~20–30 variants/sporadic subject were PCR-amplified followed by sequencing on an ABI 3130XL genetic analyser (Applied BioSystems).
To evaluate genotype–phenotype correlations in the identified genes, we gathered all intragenic published mutations from HGMD, denovo-db and current literature. We considered only missense, nonsense, canonical splice sites and exonic small indels. These variants were dichotomised into either missense/in-frame indels or LoF (out-of-frame indels, nonsense and canonical splice-site variants).
Validation of mRNA stability
RNA was extracted from blood samples (lymphocytes) with TriPure (Roche) and retrotranscribed using RevertAid H Minus First Strand cDNA Synthesis Kit (Fermentas) with random hexamers. PCR-amplification was done on cDNA using a primer pair, forward in exon 2_3 (CCACCTGGACGTATTCCACT) and reverse in exon 5 (ACTTGCCCATCTCTGGTTTC). Amplicon’s size (433 bp) was evaluated with an agarose gel, using GeneRuler 100 bp (Thermo Scientific) DNA ladder.
Results
Family 1: TP63 mutation
In family CLP-1055, we identified a 2 bp insertion (NM_003722.4: c.819_820dupCC) in exon 6 of TP63 in the father and the proband (figure 1—F1). This mutation leads to a frame-shift with a PTC (NP_003713.3: p.Gln274fs*4:). The insertion is not known in the gnomAD containing 246,134 TP63 alleles. The gene is intolerant for LoF mutations as evidenced by the PLi value of 0.98 (divergence between observed and expected counts for LoF changes) provided by ExAC. In all the 12 known TP63 coding transcripts, the frame-shift is predicted to induce PTC in an evolutionary conserved DNA binding domain (figure 2). Further, mRNA studies demonstrated that the mutant allele underwent nonsense-mediated-mRNA decay (NMD) as only the wild-type allele observed in mRNA obtained from the patient’s lymphocytes and lymphoblasts.
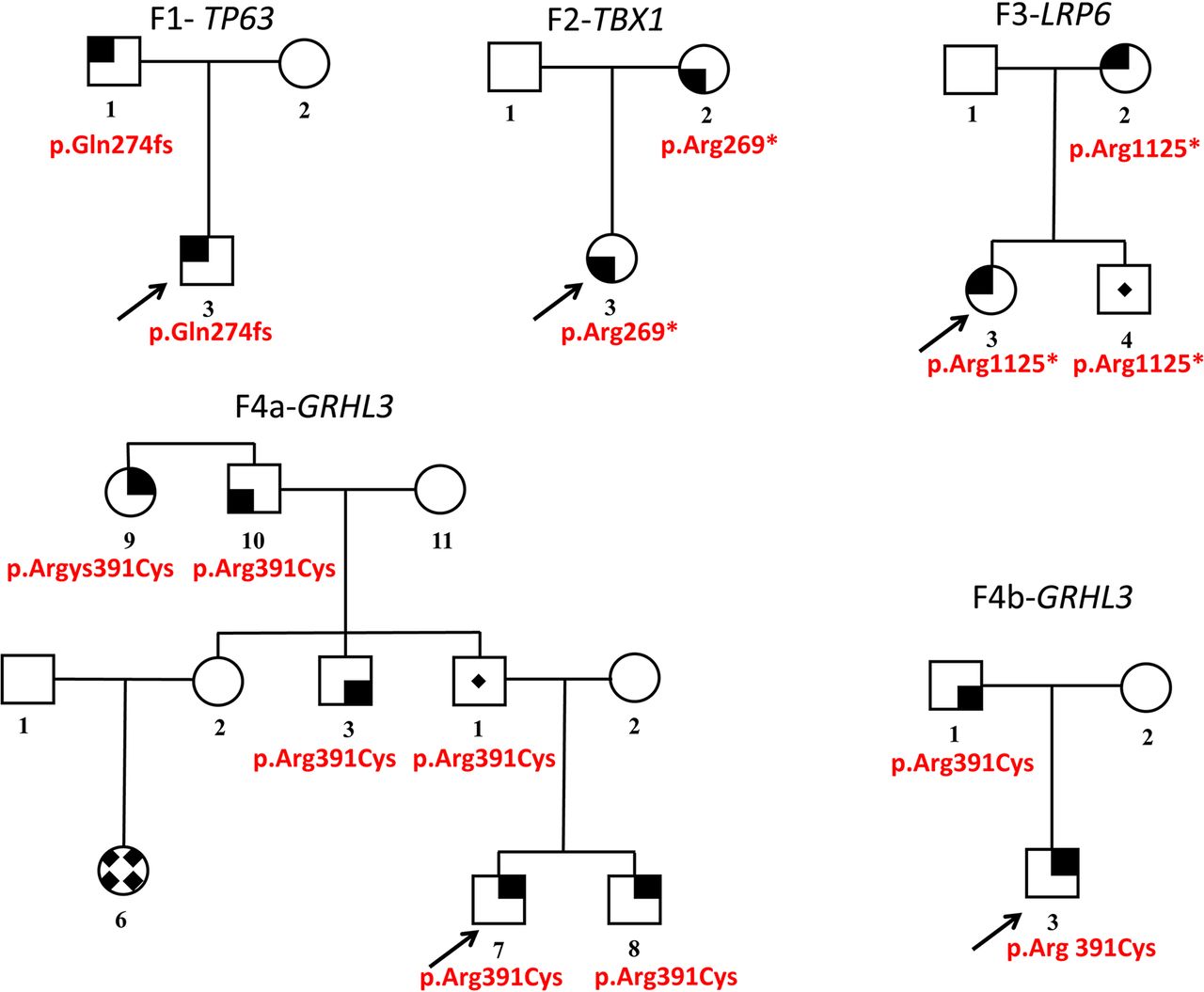
Five pedigrees with four mutations in cleft lip and/or palate (CL/P) syndrome causing genes. Family number and mutated gene marked above the pedigree (F-#- gene). Mutation is given underneath each carrier/affected. Gender symbols shaded with solid black areas indicate the subphenotype of CL/P. Upper right, CL/P; upper left, cleft palate (CP) or Pierre-Robin sequence; lower left, submucous cleft palate±bifid uvula; lower right, velopharyngeal insufficiency and/or hypernasal speech. Gender symbols with a central dot indicate carriers. A female symbol with a solid diamond pattern indicates a phenocopy CP. An arrow marks proband in each family.

TP63 protein structure; domains/motifs represented by different geometrical shapes (all scaled). ADULT, acro-dermato-ungual-lacrimal-tooth syndrome; AEC, ankyloblepharon–ectodermal defects–cleft lip/palate; DBD, DNA-binding domain; EEC3, ectrodactyly, ectodermal dysplasia and cleft lip/palate syndrome 3; ITA, inhibitor of transactivation domain; LMS, limb–mammary syndrome; RH, Rapp-Hodgkin syndrome; SAM, sterile alpha motif; SHFM, split/hand–foot malformation; TA, transactivation domain. Symbols: ✖, loss-of-function variants (exonic premature termination codon, out-of-frame indel and canonical splice-site variant); dashed line across protein, out-of-frame indel from start to end; ◉, in-frame indel; ◆, missense variants (neomutation, or present in a minimum of two affected, or functionally validated);  , other rare missense variant; dashed line across protein ending with Δ, a large 5′ deletion of several exons; number in parentheses, number of occurrences in index patients, mutations in close proximity grouped (see online supplementary table S1).
, other rare missense variant; dashed line across protein ending with Δ, a large 5′ deletion of several exons; number in parentheses, number of occurrences in index patients, mutations in close proximity grouped (see online supplementary table S1).
Supplementary file 1
The proband had a unilateral, right-sided CLP. He had no limb anomaly, no ectodermal dysplasia and no cardiac malformation. Follow-up until the age of 3.5 years showed growth and development within normal limits. His father had a unilateral, left-sided CLP. Both were re-examined for minor symptoms of TP63 disorders, yet they sustained their initial classification as nsCL/P.
Family 2: TBX1 mutation
In family CLP-678, we identified a nucleotide change (NM_080647.1: c.805C>T:) in exon 6 of TBX1 in the mother and the proband (figure 1—F2). This nonsense mutation (NP_542378.1: p.Arg269*: NP_542378.1) is located in the T-Box domain (figure 3). The substitution is unknown in gnomAD containing 277 208 TBX1 alleles. As TBX1 mRNA is not expressed in lymphocytes or lymphoblasts, mRNA stability could not be studied.
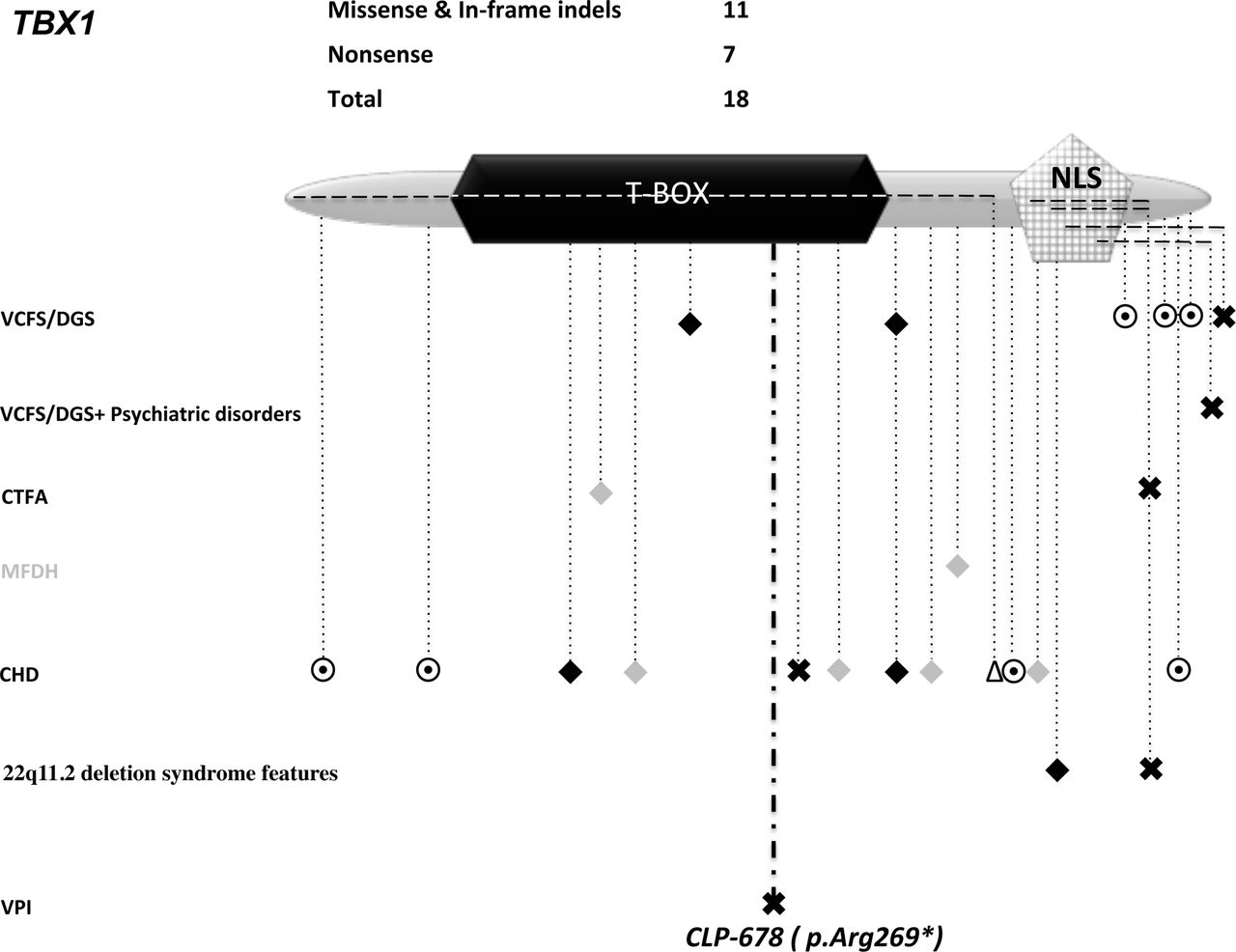
TBX1 protein structure; domains/motifs represented by different geometrical shapes (all scaled). CHD, congenital heart defect; CTFA, conotruncal face anomaly syndrome; MFDH, midline facial defects with hypertelorism; NLS, nuclear localisation signal; T-BOX, DNA-binding domain; VCFS/DGS, velocardiofacial syndrome/DiGeroge syndrome; VCFS/DGS + psychiatric disorders such as depression and Asperger syndrome; VPI, velopharyngeal insufficiency. Symbols: ✖, loss-of-function variants (exonic premature termination codon, out-of-frame indel and canonical splice-site variant); dashed line across protein, out-of-frame indel from start to end; ◉, in-frame indel; ◆, missense variant (neomutation, or present in a minimum of two affected, or functionally validated);  , other rare missense variant; dashed line across protein ending with Δ, a large 5′ deletion of several exons (see online supplementary table S2).
, other rare missense variant; dashed line across protein ending with Δ, a large 5′ deletion of several exons (see online supplementary table S2).
Supplementary file 2
The index subject was born at 35.5 weeks of gestation. She had neonatal regurgitations and bradycardia. A diagnosis of VPI with severe hypotonia of the velopharyngeal musculature was made, but the 22q11 deletion test was normal. Follow-ups showed normal development, no particular facial gestalt, yet with a persistent hypernasal speech (a consequence of VPI), despite receiving speech therapy. Apart from a neonatal patent ductus arteriosus, the index case had no cardiac structural defect.
The proband’s mother also had isolated VPI. She underwent surgery at the age of 23 years, with partial improvement. Cardiac ultrasound did not reveal any cardiac defect.
Family 3: LRP6 mutation
In family CLP-451, we identified a nucleotide change (NM_002336.2: c.3373C>T:) in exon 15 of the LDL receptor related protein 6 (LRP6) in three individuals (figure 1—F3). The substitution is not known in gnomAD containing 277 132 LRP6 alleles. This nonsense variant (NP_002327.2: p.Arg1125*:) is located in a conserved YWTD repeat in the fourth β-propeller domain (figure 4).

LRP6 protein structure; domains/motifs represented by different geometrical shapes (all scaled). ASD, autism spectrum disorder; CAD, coronary artery disease; CLP, cleft lip and palate; MtB, metabolic syndrome; nsCL/P, non-syndromic cleft lip and/or palate; NTD, neural tube defect; TA, tooth agenesis; TA+CL/P, combined tooth agenesis with CLP and mild dysmorphic features. Extracellular part: β, β propeller repeats; EGF, epidermal growth factor-like domain; LDL, LDL type A-like receptor; SP, signal peptide. Symbols: ✖, loss-of-function variants (exonic premature termination codon, out-of-frame indel and canonical splice-site variant); dashed line across protein, out-of-frame indel from start to end; ◆, missense variant (neomutation, or present in a minimum of two affected, or functionally validated);  , other rare missense variant (see online supplementary table S3).
, other rare missense variant (see online supplementary table S3).
Supplementary file 3
The proband had a BCLP with missing upper lateral incisors, and her mother had a bilateral CL. Proband’s brother is an unaffected carrier of the substitution. We could not exclude oligodontia (>6 missing teeth) in any of the three mutation carriers since the family was not available for reassessment.
Families 4 and 5: GRHL3 mutation
We identified an identical nucleotide change (NM_198174.2: c.1171C>T:) in exon 9 of Grainyhead-like transcription factor 3 (GRHL3) in two families CLP-398 (figure 1—F4a and CLP-986 (figure 1—F4b). The substitution is listed once in gnomAD containing 244 128 GRHL3 alleles. This variant leads to an arginine-to-cysteine substitution (NP_002327.2: p.Arg391Cys) in an evolutionarily conserved DNA-binding domain (figure 5). This change has been reported as a de novo variant in two patients, one with a CLP and lip pits,22 the other with spina bifida.23 In vitro and in vivo studies demonstrated a dominant negative effect.22 24

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
GRHL3 protein structure; domains/motifs represented by different geometrical shapes (all scaled). CLP, cleft lip and palate; DBD, DNA-binding domain; DD, dimerisation domain; NTD, neural tube defect; nsCP, non-syndromic cleft palate; TA, transactivation domain; VWS2, Van der Woude syndrome 2. Symbols: ✖, loss-of-function variants (exonic premature termination codon, out-of-frame indel and canonical splice-site variant); dashed line across protein, out-of-frame indel from start to end; ◆, missense variant (neomutation, or present in a minimum of two affected, or functionally validated);  , other rare missense variant;
★
, compound heterozygous mutations, identified in a single family;
★★
, homozygous mutation (see online supplementary table S4).
, other rare missense variant;
★
, compound heterozygous mutations, identified in a single family;
★★
, homozygous mutation (see online supplementary table S4).
Supplementary file 4
The proband in family CLP-398 had a Pierre-Robin sequence (PRS) (figure 1—F4a). His brother had a milder PRS. On the paternal side, an uncle had a high-arched palate with a SMCP and missing upper lateral incisors. A paternal great-aunt had a surgically repaired CP. The grandfather has hypernasal speech, according to family history. The father of the proband is an unaffected carrier.
The proband in family CLP-986 had a posterior CP (affecting the soft palate). His father had a SMCP with a bifid uvula (figure 1—F4b). Abnormalities of the lower lip, such as lip pits associated with the VWS, were excluded following detailed clinical examination in both families.
Discussion
We identified altogether 5 out of 46 index patients screened to have a clear mutation in a gene mutated in specific syndromes associated with oral clefts. The pick-up frequency among them is thus about 10% (5/46). These were all subjects with family history of nsCL/P. In total, there were 15 mutation carriers. Penetrance was high 13/15 (87%). Apart from family CLP-451, all patients were clinically re-examined for minor signs of CL/P syndromes. The assessment confirmed the isolated occurrence of CL/P. This underscores the extreme variability in expressivity in phenotypes caused by mutations in these genes, which can range from a full-spectrum syndrome to a most subtle CL/P subphenotype such as VPI.
In total, four mutations in four different genes were identified in five families (one mutation was shared by two unrelated families). The four mutations co-segregated with the oral cleft in an autosomal dominant manner in all families. They belong to the less frequent group, when known mutations are dichotomised into LoF (PTC or variant in a canonical splice site) and missense mutations. In TP63, TBX1 and LRP6, in which we identified a LoF mutation, LoF variants account for only 21/103 (figure 2), 7/18 (figure 3) and 9/28 (figure 4), respectively. In contrast, in GRHL3 (figure 5), in which we identified a missense mutation, validated missense variants23 24 account for only 7/20.
In two of the four genes, localisation of our mutations did not follow the typical grouping of mutations by type. In both TP63 and TBX1, the few known LoF mutations cluster to the 3′ end of the gene (figures 2–3 and online supplementary tables S1 and S2) and have been identified in patients with a full-blown syndrome, such as EEC3 (ectrodactyly, ectodermal dysplasia MIM #604292, and cleft lip/palate syndrome 3), ADULT (acro-dermato-ungual-lacrimal-tooth syndrome MIM #103285), RH (Rapp-Hodgkin syndrome MIM #19400), AEC (ankyloblepharon–ectodermal defects–cleft lip/palate MIM#19400), LMS (limb–mammary syndrome MIM #603543), SHFM (split/hand–foot malformation 4 MIM # 605289) or (VCFS/DGS), respectively (figures 2–3). Our LoF mutations localised to the DNA-binding domain, in the middle of the two proteins. Thus, there seems to be a genotype–phenotype correlation regarding mutation type and/or localisation for these genes.
TP63 encodes a transcription factor, which is a master regulator of epithelial lineage commitment during and after development. Heterozygous germ-line mutations in TP63 underlie at least six disorders with overlapping phenotypes, which share ectrodactyly, ectodermal dysplasia, cleft lip/palate (EEC), lacrimal duct obstruction, hypopigmentation and hyperplastic breasts and/or nipples to varying degrees. Mutations are dispersed in all functional domains of TP63, with a well-established genotype–phenotype correlation (figure 2). Penetrance is incomplete and variable expressivity is observed. EEC point mutations cluster in the DNA binding domain (DBD) and have a disruptive effect on DNA binding characteristics of all expressed isoforms. The effects of these mutations on the counterpart wild-type allele are thought to be dominant negative.25 To our best knowledge, this is a first reported indel mutation within the DBD domain. Yet, the position is not that important, as the transcribed allele undergoes mRNA decay. A larger deletion extending from exon 1 to exon 4 (encompassing most of DBD) was reported in a patient with EEC3.26 It is not known if a stable TP63 lacking most of the DBD domain is produced starting from one of downstream in-frame ATGs and what kind of activity such a short form would have.
Whether TP63’s haploinsufficiency could also be the pathomechanism for some types of TP63 disorders has been a subject of debate and was originally refuted based on inferences from mouse model phenotypes when compared with humans. Tp63−/− mice recapitulate the phenotypic spectrum observed in humans with a TP63 +/− mutation,27 while tp63−/+ mice nor patients with a 3q27del do not exhibit ectodermal defects.27 However, recent publications report patients with ectodermal dysplasia and high-arched palate,28 among other symptoms with a 3q27.3del and a variable expressivity in a CP family with 3q28del.29 This suggests that TP63 haploinsufficiency can be a driving mechanism behind clefting. Therefore, the identified TP63 LoF allele, which manifests as isolated nsCLP, suggests it to be a haploinsufficient allele.
TBX1 maps to 22q11.2, a region frequently deleted in humans (~1/4000 live births). It is associated with DiGeorge syndrome (DGS)/velocardiofacial syndrome (VCFS) (MIM 192430/188400). It regulates a number of genes via epigenetic modifications.30 31 TBX1 mutations manifest with pharyngeal arch-related developmental defects, affecting the heart, thymus and parathyroid gland. TBX1 mutations cause a recognisable spectrum of craniofacial anomalies such as CP, or VPI with typical facies and a predilection to develop neuropsychiatric disorders.
Phenotypic discordance was observed in cases with an identical deletion and also among monozygotic twins.32 Deleterious variants located within the Brachyury T domain (a well-conserved DBD, shared with T-Box family members) are associated with DGS/VCFS (figure 3 and online supplementary table S2) and some variants are associated with isolated congenital heart defects (CHDs) (figure 3 and online supplementary table S2). In vitro investigations by luciferase assay demonstrated that the only identified PTC in the DNA-binding domain of TBX1 p.Glu277*, which co-segregated in a family with CHD, is a LoF variant.33 This variant is distal to our p.Arg269* variant, which manifests as isolated VPI without any cardiac defect. There are another four reported frame-shift mutations towards the 3′ end of TBX1, all disrupting the nuclear localisation signal (figure 3). These mutants, if stable, could inhibit TBX1 dimer formation and thus have dominant-negative effects on the function of the remaining wild-type allele. The extreme variability in phenotypic expressivity and penetrance suggests that depending on the mutation’s type and localisation, they have different effects on TBX1 function, and that other, genetic co-factors play an important role.
Mutations in LRP6, a well-studied co-receptor in WNT/β-catenin-dependent signalling pathway, have a pleiotropic role in disease (figure 4 and online supplementary table S3).34 Lrp6 mimics WNT activity in enhancing neural crest formation,35 which gives rise to diverse cell lineages including cranioneural crest cells. LRP6 mutations are associated with congenital neural tube defects,36 37 and severe tooth agenesis,38 sometimes with CLP.39 The nonsense mutation we identified is located in the extracellular domain of LRP6 causing haploinsufficiency. It is located adjacent to previously identified LoF heterozygous mutations, which cause autosomal tooth agenesis with reduced penetrance. In vitro studies showed that the p.Ala19Val mutant LRP6 was retained in the endoplasmic reticulum,38 which prevented it from being visible for incoming ligands at the cell surface. This suggests that the not fully penetrant tooth agenesis phenotype is due to loss of membrane-bound LRP6 signalling. A combined phenotype (tooth agenesis and CLP) was observed in another patient with a truncating variant in the last exon of LRP6 (intracellular; figure 4). Although it is expected to escape NMD, this variant is at least partially inactive since it loses three of its five signature PPPSP motifs,40 crucial for phosphorylation of LRP6 and WNT activation. Whether mutation type and position alone, or interaction with co-factors, explain the phenotypic variability associated with LRP6 mutations remains to be seen.
GRHL3 was identified by linkage to the region 1p33–36 in a large Finnish family affected with VWS2 (MIM 606714). It was screened in a series of nsCP and VWS2 suspects. Mutations’ effects ranged from dominant negative to strong and moderate hypomorphs (figure 5 and online supplementary table S4).22 24 41 The latter mechanism loses some of its activity compared with the wild type, and as such predisposes rather than causes nsCP/VWS2. Our identified variant p.Arg391Cys has a dominant negative effect. It is associated with intriguing variability in phenotypic expressivity. As a de novo mutation, it manifested with CLP and lip pits in one Filipino subject, while with isolated spina bifida in a second subject. We identified it as a germline mutation in two families of Belgian descent, both of which display a remarkable intrafamilial and interfamilial variability varying from PRS and CP to hypernasal speech (figure 1—F4a–F4b). Given that this mutation is a C>T transition, occurring in a CpG dinucleotide known to easily undergo deamination, this mutation likely represents a mutational ‘hot-spot’. The occurrence in different ethnicities underscores this.
Lip pits, a hallmark of VWS, with an approximate penetrance of 80%, is observed in about 30% of patients with a GRHL3 mutation.22 41 However, lip pits were not present in either one of our families contrary to the aforementioned patient with VWS2. Although mild micrognathia was described, PRS observed in the proband and his sibling in family CLP-398 is novel and demonstrates once more the important clinical variability associated with variants in oral cleft ‘syndrome-causing’ genes, and the necessary quest to investigate modifiers.
Our results, as well as previously published data including IRF6,20 42 MSX1,43 TBX22 44 and GRHL3,41 substantiate evidence for Mendelian transmission (with incomplete penetrance) of aetiological variants in ‘syndrome-causing genes’ in subclinical or non-syndromic cleft families. Therefore, custom-designed panels including CL/P-syndrome-causing genes should be used in diagnostic screens when a particular syndrome is suspected and also for families with nsCL/P. The benefits are dual; this will improve genetic counselling (by providing more informative chances of inheritance of 25%–50% versus an empiric of 4%–17%) and phenotyping of the patients (in search of minor/overlooked symptoms relevant to the syndrome) and also expand our understanding of molecular mechanisms underlying CL/P.
Such diagnostic screens will deliver many rare variants that will stratify CL/P cohorts more uniformly. Using genomic technologies such as WES or WGS (whole genome sequencing) on stratified cohorts will subsequently facilitate the teasing out of genetic modifiers such as coding variants in other genes, distantly acting enhancers, promoters and other conserved non-coding regions. Another unexplored explanation for sporadic CL/P are somatic mutations in developmentally relevant tissues in one of the known syndrome-linked genes. Proof of this concept has already elegantly been shown for various sporadically occurring vascular anomalies.45 46 Unfortunately, targeted exomes and/or genomes of relevant tissues to CL/P are not that feasible to perform, until we determine the exact relevant tissues.
Concluding remarks
When syndromes manifest at the mild end of their spectra, they are often overlooked by clinicians. Our study clearly demonstrates the need to broaden criteria for diagnostic genetic screens to include patients with nsCL/P with a family history, as in 10% of them a causative Mendelian mutation could be identified. It could be that in an important percentage of the remaining cases, the same genes are involved due to an indel or copy number alteration (CNVs) not easily detectable by WES. WGS will therefore likely be the best choice for diagnostic screens of families with a history of nsCL/P.
References
Footnotes
Contributors MB and MV prepared the manuscript. MB performed patient selection for WES, WES data analysis and validation/co-segregation of identified variants. BD, NR and BB were in charge of enrolment of subjects and collecting samples. RH helped in bioinformatic analyses of WES data. BD, NR, ST, SBS, OB, BD, GF and BB collected the clinical data. All authors contributed to re-drafting of the manuscript. MV conceived and coordinated the project and is responsible for the overall content. All authors have seen and approved the final manuscript.
Funding These studies were partially supported by funding from the Belgian Science Policy Office Interuniversity Attraction Poles (BELSPO-IAP) programme through the project IAP P7/43-BeMGI; BridgeIris RBC/2013-PFS-EH-11; the Fonds de la Recherche Scientifique—FNRS; CdR: J.0080.16 (all to MV). We also acknowledge the support of la Communauté française de Wallonie-Bruxelles, la Lotterie Nationale, Belgium and la région Haut de France, France. The authors thank the Genomics Platform of Université catholique de Louvain for next-generation sequencing analyses and the Foundation against Cancer, Belgium.
Competing interests None declared.
Patient consent Not required.
Ethics approval This study obtained ‘La Commission d’Ethique Biomédicale Hospitalo-Facultaire’ approval (2015/02NOV/572) by the medical faculty at University of Louvain, Brussels, Belgium.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All data relating to the study are available within the manuscript and accompanying online supplementary files.