Article Text

Abstract
Background A substantial amount of nuclear genes have been identified to be implicated in genetic hearing loss, while X-linked hearing loss is genetically heterogeneous and relatively infrequent.
Objective To identify the causative gene mutation in a five-generation Chinese family with an X-linked recessive syndromic hearing loss (SHL).
Methods Targeted X-chromosome exome sequencing was conducted, and cosegregation analysis was performed in the members of the affected family. The in silico and expression studies were also performed.
Results A 2-bp missense mutation (c.1717_1718GC>AA, p.A573N) in the G protein-coupled receptor associated sorting protein 2 (GPRASP2) gene was identified in four hemizygous male patients and two heterozygous female carriers, which was cosegregated with the clinical phenotypes in this family. In silico analysis supported that this gene mutation is functionally deleterious, and it was detected that homologous Gprasp2 was highly expressed in multiple structures of the mouse cochlea, which suggested that GPRASP2 might be the genetic cause for the described disease phenotypes.
Conclusions This study presented a novel X-linked SHL combined with unique and unrecognised clinical features, and a missense variation of GPRASP2 was first identified to be implicated in X-linked SHL.
- Genetics
- Genetic screening/counselling
- Molecular genetics
- Clinical genetics
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Hereditary hearing loss accounts for at least half of cases with congenital or prelingual-onset hearing loss, which is associated with genetic mutations in autosomal dominant, autosomal recessive, maternal and X-linked inheritance patterns.1–4 An impressive amount of nuclear genes have been implicated in genetic hearing loss, while X-linked hearing loss is genetically heterogeneous and relatively infrequent, accounting for 1%–5% hereditary hearing loss.4 To date, only four X-linked genes have been described for non-syndromic hearing loss (NSHL): POU3F4 (OMIM 300039),5 PRPS1 (OMIM 311850),6 SMPX (OMIM 300226)7 ,8 and COL4A6 (OMIM 303631).9 Hearing loss could also be one of the clinical features in 11 human genetic syndromes (http://hereditaryhearingloss.org/) and some mitochondrial diseases.10 Those syndromes are commonly caused by mutations in a single gene or multiple genes. Among these causative gene mutations for syndromic hearing loss (SHL), only one X-linked gene, COL4A5 (OMIM 301050), was identified to be implicated in SHL as one of the symptoms of Alport syndrome (OMIM 301050).11
Here we describe a Chinese family with a unique and previously unrecognised combination of clinical features, including ear anomalies, congenital conductive or mixed hearing loss and facial dysmorphism with bilateral ptosis. All affected familiar members were male, whereas the female subjects are phenotypically normal, which highly suggested the X-linked recessive inheritance of this affected family. Based on mentioned above, targeted X-chromosome exome sequencing (XES) was conducted to identify the underlying genetic defect. Via cosegregation analysis of the affected family, we identified a novel candidate X-linked SHL-related gene, encoding G protein-coupled receptor associated sorting protein 2 (GPRASP2, NC_000023.11, NM_001184874.2, Xq22.1). In silico and expression studies also strongly supported the implication of GPRASP2 in hearing function.
Materials and methods
Subjects and clinical investigation
A general overview of the family's pedigree is presented in figure 1A. The pure tone audiograms test showed that the proband (V-4) exhibited bilateral symmetric and conductive hearing loss (see online supplementary figure S1 and table S1), while the parents (IV-11, IV-12) both presented normal hearing. A detailed medical history, including age at onset, evolution of hearing loss, presence of tinnitus, pathologic changes in the ear (or other relevant clinical manifestations), medication, noise exposure and family history, was interviewed in the family. A comprehensive examination of this family was conducted, including clinical examinations, audiological tests, intelligence evaluation, development evaluation, high-resolution CT scan of the temporal bone and laboratory tests. Three hundred normal hearing individuals were also included as ethnic-matched controls. This study was approved by the ethics committee of Nanjing Medical University, Jiangsu province, and informed consents were obtained from the participants.

Clinical features and mutational analysis of the affected family with X-linked syndromic hearing loss (SHL). (A) Pedigree of affected family with X-linked SHL. Filled symbols for males (squares) and females (circles) represent patients, and empty, unaffected ones. An arrow denotes the proband (V-4). (B) Frontal and lateral views of affected family members (V-4, IV-9, III-6 and III-13). (C) CT scans of the temporal bone in selected patients. III-6: showing bulbous dilatation of internal auditory canal (IAC) with incomplete separation of cochlea basal turn from the fundus of IAC; III-13: showing narrow (right ear) and atresic (left ear) auditory canals; V-4: displaying normal inner ear. (D) Mutational analysis in the family members. In available 14 family members, there were 4 patients and 2 carriers (high lightened in red), and the mutation was not found in the remaining 8 family members with normal phenotypes.
supplementary figures and tables
Targeted X-chromosome capture and exome sequencing
Two affected individuals (III-13, IV-9) and one normal hearing family member (III-4) were included in targeted XES. A minimum of 3 μg DNA was used to make the indexed Illumina libraries according to the manufacturer's protocol. The final library size 300–400 bp including adapter sequences was finally selected. The whole exon regions of the X-chromosome were enriched using the target enrichment kit (MyGenostics, Beijing, China). The enrichment libraries were sequenced on Illumina HiSeq 2000 sequencer (Illumina, San Diego, California, USA) with 100 bp paired-end reads, and it was ensured that each sample was covered to an average sequencing depth of at least 80-fold.
Illumina clean reads were aligned to human reference genome using the Burrows-Wheeler Aligner (BWA) programme and quality scores were recalibrated and realigned to reference using the GATK software package. We focused on non-synonymous coding substitutions and frameshift mutations on X-chromosome. The identified variant should be shared by the three included individuals and cosegregated with the syndromic phenotypes in these three subjects. The possible pathogenic variant was evaluated according to the evolutionary conservation and change of conserved amino acid.
Mutation validation and in silico analysis
PCR primers flanking the GPRASP2 exons and exon–intron boundaries are presented in online supplementary table S2. The whole exons of GPRASP2 from all the available family members and 300 ethic-matched controls were subjected to the mutational analysis by direct PCR and Sanger sequencing.
The conservation analysis of residues in a multiple sequence alignment was conducted using Clustal X software. The three-dimensional structure of GPRASP2 was modelled based on the lowest energy using Robetta programme (http://robetta.bakerlab.org/). The structure of mutant protein was built by the Swiss-Model tool (http://swissmodel.expasy.org/) which generates modelling for the target protein based on a sequence alignment between the target protein and a suitable template structure.
Gprasp2 expression analysis in mice
Total RNA of eight tissues (the cochlea, liver, kidney, muscle, lung, spleen, heart and brain) was obtained from a 12-week mouse (C57BL/6). The primers used for the amplification of Gprasp2 cDNA (NM_001163015.1) were provided in online supplementary table S3. The reverse transcription was performed according to the manufacturer's protocol (Invitrogen, Carlsbad, California, USA), and the product cDNA was used to determine the mRNA expression levels by reverse transcription-polymerase chain reaction (RT- PCR) (Applied Biosystems, Step One Plus, USA).
The immunohistochemical study on the mouse cochleae was performed as mentioned in our previous work.12 Briefly, rabbit anti-Gprasp2 antibody (Proteintech, USA) was used as the primary antibody (1:80), and horseradish peroxidase (HRP) conjugated goat anti-rabbit IgG (Santa Cruz, USA) was used as the secondary antibody (1:100).
Results
Clinical assessment
The pedigree of the family includes 60 members spanning five generations (figure 1A). All six affected individuals were male; four were alive and diagnosed with congenital, hereditary hearing loss by audiological evaluation or collected information of medical history. Detailed clinical features in these subjects are summarised in online supplementary table S1 and figure S1. In otological examination, abnormal auricular shapes including bat ear (III-13), posteriorly rotated small ear (IV-9), incomplete antihelix (IV-9, V-4), antitragus formation (IV-9, V-4) and bilateral ptosis (III-13, IV-9 and V-4) were found in affected cases (figure 1B). In addition, the imaging study of the temporal bone demonstrated that those patients who exhibited bilateral mixed hearing loss had also a similar inner ear malformation: abnormally dilated internal auditory canal (IAC) with or without incomplete separation of cochlea basal turn from the fundus of IAC (figure 1C).
XES data and mutation validation
An average of 492.06 Mb of raw data was generated with at least 100X average coverage for each individual as paired-end 100 bp reads (see online supplementary figures S2, S3 and table S4). We identified an average of 372 SNPs (synonymous and non-synonymous) in coding regions (see online supplementary table S5). A total of eight variants were screened after comparison with SNP and indels databases (see online supplementary tables S6 and S7). Among them, five variants (including three non-synonymous SNPs and two indels) were predicted to potentially have a functional impact on the genes (see online supplementary tables S8–S10). In addition, it strongly indicated that a 2-bp missense mutation of c.1717-1718GC>AA in exon 5 of GPRASP2 could be the most likely candidate pathogenic variant, which was confirmed by cosegregation validation via Sanger sequencing subjected to all candidate variants.
The cosegregation analysis showed that all the normal hearing family members are either unaffected (eight individuals) or c.1717-1718GC>AA heterozygous carriers (III-4, IV-11), while a hemizygous mutation was detected in III-6, III-13, IV-9 and V-4 in this pedigree (figure 1D). The patient IV-9 and V-4 carried a hemizygous mutation of c.1717-1718GC>AA, which was inherited from the heterozygous mothers of III-4 and IV-11, respectively, who had normal hearing. Additionally, it was noteworthy that a highly skewed X-chromosome inactivation was detected in III-4 and IV-11 (see online supplementary files (X-inactivation )). Thus, the mutation of c.1717-1718GC>AA in GPRASP2 was completely cosegregated with the X-linked recessive hearing loss phenotype of this family. Thereafter, we sequenced the entire coding region and exon–intron boundaries in GPRASP2, and no other mutations in GPRASP2 were identified in any available family members. No mutation of c.1717-1718GC>AA in GPRASP2 was detected in 300 ethnicity-matched controls.
supplementary file
In silico and expression analysis
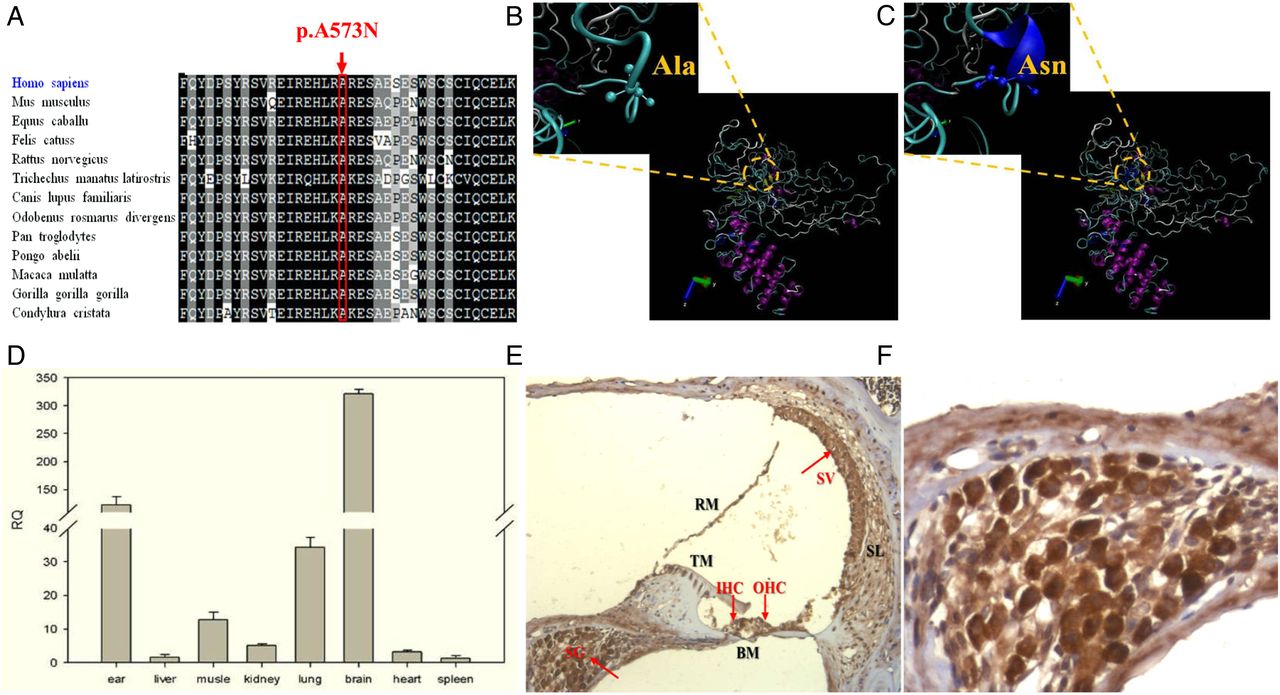
The mutation of c.1717-1718GC>AA in GPRASP2 caused a change of amino acid corresponding to codon 573 (p.A573N), which is highly conserved in different species by Clustal X Software (figure 2A), and the structural modelling revealed that the p.A573N mutation could result in the steric hindrance and polarity alternation of side chain, and lead to the structural change of protein (figure 2B, C), which might impair the function of GPRASP2.
{kind=link}
{kind=link}
Expression and in silico analysis of GPRASP2/Gprasp2. (A) Multiple alignment of GPRASP2 amino acid sequences in different species. (B) Structure modelling of normal GPRASP2. (C) Structure modelling of mutated GPRASP2 of p.A573N (the structure changes were emphasised by yellow circle). (D) RT-qPCR analysis of the Gprasp2 expression in mouse tissues. (E and F) Immunohistochemical localisation of Gprasp2 in the mouse cochlea (The stained slides were observed under the Leica upright microscope (Leica DM 4000B, Germany). (E) Low magnification (10×): Gprasp2 was strongly expressed in the spiral ganglion (SG), stria vascularis (SV), inner hair cells and outer hair cells. Gprasp2 was also detected in the spiral ligament. (F) High magnification (40×): SG and SV showed intensively staining of some positive cells).
The Gprasp2 gene was strongly expressed in the mouse brain and cochlea, posteriorly in lung and muscle tissues (figure 2D), which was consistent with the previous studies.13 The immunohistochemical study on the mouse cochlea was performed to localise Gprasp2 in multiple structures of the mouse cochlea. Intensive staining for Gprasp2, which densely packed cytoplasmic structures of mostly positive cells, was detected in the spiral ganglion, stria vascularis, spiral ligament, inner hair cells and outer hair cells (figure 2E, F).
Discussion
In this study, a novel SHL-associated candidate gene, GPRASP2, was identified in a Chinese family with X-linked SHL using XES. The mutation of c.1717_1718GC>AA (p.A573N) in exon 5 of GPRASP2 was identified to be completely cosegregated with the clinical phenotype of hearing loss in this family and absent in the controls. In silico and expression analysis strongly suggested that the mutation of GPRASP2 could be functionally deleterious and might lead to the pathogenicity, and thereby the disease phenotype. To our knowledge, since the X-linked COL4A5 gene was identified to be implicated in SHL as one of the symptoms of Alport syndrome,11 the GPRASP2 gene could be the second X-linked gene associated with SHL.
GPRASP2, also known as GASP2, consists of a single coding exon (exon 5) and encodes an 838 amino acid residue protein which is a member of the G protein-coupled receptor associated sorting proteins (GASPs) family.14 ,15 The GASP family including GASP1–GASP10 plays a role in G protein-coupled receptors (GPCRs) sorting. All the GASP family members display sequence similarities in their carboxyl-terminal domains of 250 amino acids residues, and GASP1–GASP5 contain a repeated motif of 15 amino acids outside this domain.15 It was noteworthy that the p.A573N mutation was just located in the carboxyl-terminal domain, which indicated that the carboxyl-terminal domain could play an important role in GPRASP2 functioning, and the mutation in the location could have a functional impact on the gene.
Tissue distribution of ESTs indicates that GPRASP2, like most of other GASP family members, is predominantly expressed in the central nervous system.14 ,15 Additionally, strong expression of the Gprasp2 gene was detected in the cochlea of 9-week mouse using a microarray assay (http://research.meei.harvard.edu/Otopathology/tbimages/mouse.html), which was consistent with the results in our study. Cytological and subcellular study showed that GPRASP2 is found to be expressed in the cytoplasm and cell membranes of undifferentiated cells as well as in newly formed neurite-like extensions of retinoic acid-induced differentiated cells.13 In vitro binding studies demonstrated that GPRASP2 had the potential to interact with the carboxyl-terminal tail of several GPCRs including β-1 and β-2 adrenergic receptors,16 growth and differentiation factor-11,17 calcitonin and dopamine D2 receptors,18 indicating that the GPRASP might serve as general adaptors important for GPCRs' turnover. These interactional proteins are involved in a wide variety of processes including carcinogenesis, control of cellular ageing and survival, regulation of circadian rhythm and lysosomal sorting of GPCRs. Horn et al19 investigated the interaction between huntingtin and GPRASP2, and suggested that huntingtin protein might affect the receptor trafficking via the interaction with GPRASP2 which could cause the Huntington's disease. However, no functional assessments of GPRASP2 variants producing the X-linked syndromes and deafness phenotype were previously reported until it was identified in our work. It was presumed that the p.A573N mutation might affect its interactions with GPCRs, impair the function of GPCRs which could transduce diverse extracellular signals to intracellular effectors modulating cell function,20 lead to the pathogenicity and present SHL phenotypes.
In summary, we reported GPRASP2 as a novel gene mutated in an X-linked SHL. Although this SHL-causative mutation is now only detected in a Chinese family case, our study had strongly suggested that the GPRASP2 might play a significant role in the hearing regulation and occurrence of this novel X-linked syndrome. However, it is still necessary to be validated in other familiar or sporadic cases, and the function of GPRASP2 gene as well as the GPRASP2–proteins interactions will be investigated to illuminate the pathogenic mechanisms involved in this novel X-linked SHL in our future work.
Acknowledgments
The authors would like to thank the family members for their invaluable cooperation and participation. The authors thank MyGenostics Co. (Beijing) for targeted XES. The authors are grateful to Dr Kui Meng for helping the immunohistochemical analysis.
References
Footnotes
GX and JY contributed equally.
Correction notice This article has been corrected since it published Online First. Non-syndromic hearing loss was incorrectly abbreviated to SHL, and SHL was not expanded in the first instance. Both these instances have been updated.
Contributors The first two authors (GX and JY) contributed equally to this work. GX, JY, QW, XQ and LW: performed the experiments. CL, XQ and YL: contributed reagents/materials/analysis tools. GX and JY: wrote the paper. XC: conception and design, revised the article.
Funding This research was supported by the grants from the National Natural Science Foundation of China (31171217, 31571302) and the Key Research and Development Program of Jiangsu Province (Social Development: BE2016762) to XC; the grants from Jiangsu Health Administration of China (LJ201120) and the Research Special Fund for Public Welfare Industry of Health, Ministry of Health of China (201202005) to GX.
Competing interests None declared.
Patient consent Obtained.
Ethics approval The Ethics Committee of Nanjing Medical University.
Provenance and peer review Not commissioned; externally peer reviewed.