Article Text

Abstract
Background Osteonecrosis of the femoral head is a debilitating disease that involves impaired blood supply to the femoral head and leads to femoral head collapse.
Methods We use whole-exome sequencing and Sanger sequencing to analyse a family with inherited osteonecrosis of the femoral head and fluorescent Ca2+ imaging to functionally characterise the variant protein.
Results We report a family with four siblings affected with inherited osteonecrosis of the femoral head and the identification of a c.2480_2483delCCCG frameshift deletion followed by a c.2486T>A substitution in one allele of the transient receptor potential vanilloid 4 (TRPV4) gene. TRPV4 encodes a Ca2+-permeable cation channel known to play a role in vasoregulation and osteoclast differentiation. While pathogenic TRPV4 mutations affect the skeletal or nervous systems, association with osteonecrosis of the femoral head is novel. Functional measurements of Ca2+ influx through mutant TRPV4 channels in HEK293 cells and patient-derived dermal fibroblasts identified a TRPV4 gain of function. Analysis of channel open times, determined indirectly from measurement of TRPV4 activity within a cluster of TRPV4 channels, revealed that the TRPV4 gain of function was caused by longer channel openings.
Conclusions These findings identify a novel TRPV4 mutation implicating TRPV4 and altered calcium homeostasis in the pathogenesis of osteonecrosis while reinforcing the importance of TRPV4 in bone diseases and vascular endothelium.
- TRPV4
- osteonecrosis of the femoral head
- novel mutation
- calcium channel
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Osteonecrosis of the femoral head is characterised by compromised blood flow to the femoral head leading to bone cell apoptosis and its eventual collapse. The aetiology and pathogenesis of the disease are not fully elucidated, with treatment at advanced stages involving total hip arthroplasty. However, microvascular thrombosis is a common finding in non-traumatic osteonecrosis, indicating a vascular role in disease pathogenesis.1 A major risk factor for non-traumatic osteonecrosis includes high-dose glucocorticoid treatment for inflammatory disorders and haematological malignancies, a complication of glucocorticoid treatment.2 Inherited osteonecrosis and osteonecrosis in the absence of risk factors are rare, with the only previously identified genetic cause of inherited osteonecrosis of the femoral head consisting of three mutations in COL2A1, which expresses type II collagen and is essential to cartilage.3 ,4
Transient receptor potential vanilloid 4 (TRPV4) forms a Ca2+-permeable cation channel that is stimulated by heat, cell swelling, mechanical stress, and endogenous and synthetic ligands.5 In the small arteries that regulate systemic resistance, endothelial TRPV4 channels regulate blood flow by controlling endothelium-dependent vasodilation in response to physiological agonists.6 ,7 At the bone level, TRPV4 promotes terminal differentiation and inhibits apoptosis of bone-absorbing osteoclasts.8 Mutations in TRPV4 are pleiotropic with over 50 mutations pathogenic for six skeletal dysplasias (brachyolmia type 3, spondylo-epimetaphyseal dysplasia Maroteaux pseudo-Morquio type 2, spondylometaphyseal dysplasia Kozlowski type, parastremmatic dysplasia, metatropic dysplasia, familial digital arthropathy brachydactyly) and three motor and sensory neuropathies (congenital distal spinal muscle atrophy, Charcot–Marie–Tooth disease type 2C and scapuloperoneal spinal muscular atrophy).5
Methods
Study approval
All participants provided written informed consent for their involvement. The research protocol was approved by the McGill University Institutional Review Board. All participants of the sporadic osteonecrosis cohort provided written informed consent for DNA analysis relating to osteonecrosis studies.
Clinical and genetic analyses
Clinical analyses involved complete physical examination including growth parameter measurements, hip radiography, total body skeletal surveys, MRI and nerve conduction studies. Detailed clinical information is provided in the online supplementary note. Blood samples were obtained from affected family members to evaluate for thrombophilia markers, as described previously.9 Blood samples or buccal smears were obtained from all affected and one non-affected family members for genomic DNA (gDNA) and processed as previously described.9 Candidate genes were identified by whole-exome sequencing and the TRPV4 mutation was verified by segregation analysis.
Supplemental material
Cell culture and expression studies
Dermal fibroblasts were obtained from the proband and a healthy 23-year-old female control. HEK293 cells were transduced to stably overexpress wild type or mutant TRPV4 with a C-terminal FLAG. Expression was determined by anti-FLAG western blotting against whole-cell extracts of transduced HEK293 cells. TRPV4 mRNA expression was determined in fibroblasts and transduced HEK293 cells by qPCR.
Ca2+ imaging and analysis
Ca2+ signals were imaged in Fluo-4, Acetoxymethyl (AM) ester-loaded cells perfused with HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) physiological salt solution at room temperature by confocal fluorescence microscopy at 30 frames/s.6 ,7 Images were analysed using custom-designed software (A. Bonev University of Vermont, USA).
Statistics
Statistical significance of qPCR and densitometry results was determined using Student's t-test where appropriate. Statistical analysis of Ca2+ imaging was performed using OriginPro 7.5. Data are mean±SEM. Student's t-test and one-way analysis of variance with post hoc Bonferroni tests were used for comparisons between two groups and among more than two groups, respectively. p Values <0.05 were considered significant.
Detailed methods are available in the online supplementary methods.
Results
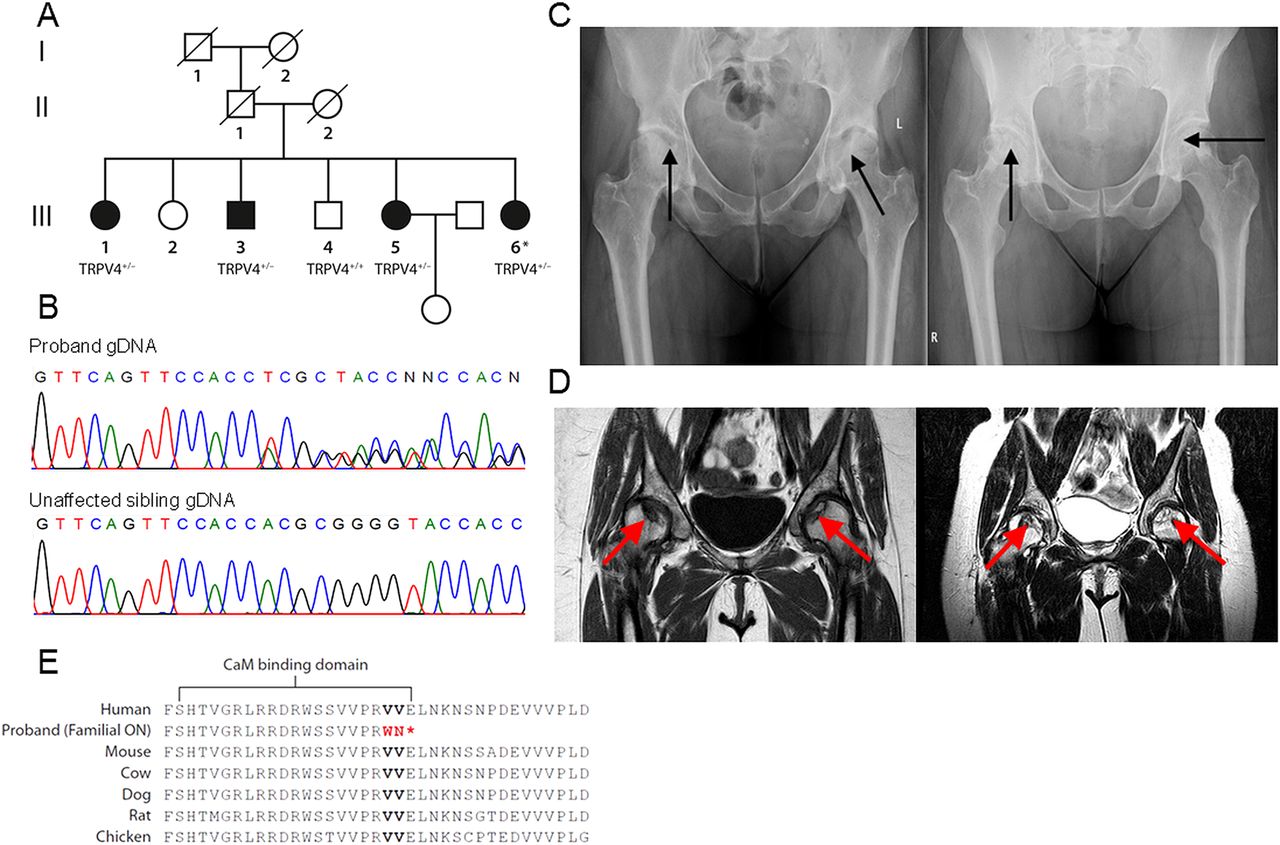
The proband was diagnosed with osteonecrosis of the femoral head at age 21 and in the same year had bilateral hip core decompressions and bone grafting. There was a positive family history with four of six siblings identified with advanced bilateral osteonecrosis of the femoral head (figure 1A), all harbouring a heterozygous mutation in TRPV4 (figure 1B) and absence of risk factors. Diagnosis was made by several bone radiologists and orthopaedic surgeons using modalities including radiography (figure 1C) and MRI (figure 1D), the gold standard for diagnosis. The extent of hip involvement or staging was evaluated according to the Steinberg classification.10 Osteonecrosis was ruled out in one unaffected sibling by radiography (see online supplementary figure S1) and the other unaffected sibling did not participate in the study. Parents and grandparents are deceased; family members recall symptoms of joint pain in their father that were never evaluated. TRPV4-associated skeletal dysplasias were ruled out by a complete physical examination performed by a medical geneticist and total body skeletal surveys. Characteristics of these skeletal dysplasias including short stature, platyspondyly, abnormal vertebral hooking or beaking or osteoarthropathy of the fingers and toes were absent (see online supplementary note for clinical details). Classical osteoporosis was also absent. TRPV4-related polyneuropathies were ruled out by neurological examination and motor and sensory nerve conduction studies performed by a neuromuscular neurologist. Thrombophilia markers, implicated as causative factors in osteonecrosis, were negative in affected family members (see online supplementary table S1).11 When the clinical profiles of the affected family members were compared with a cohort comprising 49 patients with sporadic osteonecrosis of the femoral head described previously (see online supplementary table S2), wild type for the TRPV4 mutation (see online supplementary figure S2) and absent for thrombophilia markers, all affected family members had bilateral hip involvement, whereas only two-thirds of the sporadic patients had bilateral disease.9 Also, approximately 90% of these patients had one or more risk factors.

Clinical and genetic findings in a family with inherited osteonecrosis of the femoral head. (A) Family pedigree. Siblings affected with osteonecrosis of the femoral head (black symbols) all harbour a heterozygous frameshift deletion in transient receptor potential vanilloid 4 (TRPV4) (TRPV4+/−). In unaffected siblings (white symbols) TRPV4 was wild type in one unaffected sibling (TRPV4+/+) and the other unaffected sibling did not participate in the study. One affected sibling has an unaffected 5 year old daughter. Parents and grandparents are deceased. (B) Sequencing shows the heterozygous TRPV4 mutation in proband (III-6) gDNA, wild type in unaffected sibling III-4. (C) Radiographs of the pelvis of two affected siblings showing femoral head changes (black arrows). Using the Steinberg classification, sibling III-5, left, has Stage V disease of left hip and Stage IV disease of right hip. The proband (III-6), right, has Stage IV disease of left hip and Stage III of right hip. (D) MRI images of the pelvis corresponding to radiographs showing femoral head changes (red arrows). Sibling III-5, left, has Stage V disease of the left hip and Stage IV disease of right hip. The proband (III-6, right) has Stage IV disease of both hips. (E) Sequence conservation of TRPV4 C-terminal amino acid residues. The altered residues (red) and stop codon (*) are indicated in family members affected with osteonecrosis (ON). Altered residues reside within a calmodulin-binding domain.12
Genetic analysis involved first excluding the common COL2A1 mutation in the proband followed by exome sequencing of proband genomic DNA (see online supplementary figure S3). No mutations in COL2A1 were present. Variant filtering focused on mutations in collagen genes and potentially damaging mutations (see online supplementary figure S4). Three candidate genes were identified: two mutations in collagen genes (COL12A1:c.G3532A and COL5A1:c.C260A) and a novel c.2480_2483delCCCG frameshift deletion followed by a c.2486T>A substitution in one allele of TRPV4 (NM_021625.4), included due to its bone-related phenotypes and the damaging nature of the mutation (see online supplementary table S3). Segregation analysis of the candidate genes confirmed the TRPV4 mutation in all affected siblings, wild type in the unaffected sibling (see online supplementary table S4 for primer sequences). The affected siblings did not carry the two collagen gene variants identified from exome sequencing and they were therefore eliminated as candidate genes. The TRPV4 mutation is located in the last exon and was confirmed in proband cDNA (see online supplementary figure S5). It truncates the 871 amino acid protein (V829WfsX3), eliminating a highly conserved region including residues within a calmodulin-binding domain (figure 1E).12 While all previously identified pathogenic TRPV4 mutations are autosomal dominant, only two other mutations are truncating and no other mutations are located beyond residue 799 (see online supplementary figure S6).
To measure the activity of wild-type and mutant TRPV4, HEK293 cells were transduced to stably overexpress wild-type or mutant TRPV4 (TRPV4-WT and TRPV4-V829WfsX3, respectively) with a C-terminal FLAG tag. Human-derived dermal fibroblasts obtained from the proband and a healthy control were also measured. It was not possible to obtain tissue from the disease site in affected family members. TRPV4 mRNA expression was confirmed in fibroblasts and transduced cells by qPCR (see online supplementary figures S7A and S7B). Anti-FLAG western blot confirmed wild-type and mutant TRPV4 expression in TRPV4-WT and TRPV4-V829WfsX3 cells, respectively (see online supplementary figure S8A), with twofold greater expression in TRPV4-WT cells (n=4, online supplementary figure S8B). Upper bands were confirmed as glycosylated TRPV4 by PNGase F deglycosylation (see online supplementary figure S9).13 Anti-TRPV4 antibodies could not detect TRPV4 in fibroblasts, likely due to low TRPV4 expression relative to TRPV4-WT and TRPV4-V829WfsX3 cells determined by qPCR (see online supplementary figure S10).
We recorded Ca2+ influx through TRPV4 channels within a cluster in the form of TRPV4 Ca2+ sparklets with image analysis providing indirect measurement of single channels.6 The opening of TRPV4 channels at each sparklet site causes a local increase in fluorescence that is step-like or quantal, representing the opening of one channel from the cluster. In the fibroblasts, baseline TRPV4 sparklet activity per site, calculated as NPO (where N is the number of channels per site and PO is the probability of finding the channel being open),7 was low, but significantly higher in proband versus control fibroblasts (n=5, p=0.0264; data not shown). An all-points histogram of fractional fluorescence (F/F0) traces revealed a quantal (single channel) level of 0.28 ΔF/F0 (see online supplementary figure S11), similar to previous measurements in native endothelial cells.6 We stimulated channel activity with TRPV4 agonist GSK1016790A (GSK101 hereafter, 3 nM) and found that sparklet activity per site was 3.3-fold higher in proband fibroblasts versus control (figure 2A). However, the observed maximum number (N) of channels per site, determined by dividing the maximum fluorescence signal by the quantal level (0.28 ΔF/F0), was four for both control and proband fibroblasts, suggesting that higher sparklet activity (NPO) was not due to more channels. Measurement of sparklet activity in native HEK293 cells identified no discernible sparklet activity in the absence or presence of GSK101 (1–100 nM). In the TRPV4-WT and TRPV4-V829WfsX3 cells, baseline activity was high, with 1 nM GSK101 causing Ca2+ overload and cell death. However, addition of selective TRPV4 inhibitor GSK2193874 (GSK219; 100 nM) almost completely inhibited (93%) sparklet activity (see online supplementary figure S12). The baseline TRPV4 sparklet activity per site was 2.5-fold higher in TRPV4-V829WfsX3 vs TRPV4-WT cells (figure 2B) despite a maximum of four channels per site, as seen in fibroblasts, and lower relative TRPV4 expression in TRPV4-V829WfsX3 cells, as determined by western blot. These functional TRPV4 measurements in fibroblasts and transduced HEK293 cells identify a novel TRPV4 gain-of-function mutation.

{kind=link}
{kind=link}
Longer transient receptor potential vanilloid 4 (TRPV4) channel open times increased the channel activity in proband fibroblasts and mutant TRPV4-transduced HEK293 cells. The TRPV4 channel activity was recorded optically as TRPV4 sparklets as described previously6 ,7 in Fluo-4 AM-loaded TRPV4-transduced HEK293 cells and fibroblasts in the presence of cyclopiazonic acid (CPA, SERCA inhibitor) at room temperature. Non-transduced HEK293 cells did not show any TRPV4 sparklet activity in the presence of TRPV4 channel agonist GSK1016790A (1–100 nM). (A) A greyscale image of control fibroblasts, left; representative fractional fluorescence (F/F0) traces from the control and proband fibroblasts, middle, with dotted lines representing the quantal levels derived from the all-point histograms (see online supplementary figure S11); and a bar graph comparing TRPV4 channel activity per site in control and proband fibroblasts, right. Data are mean±SEM (n=15–28 sparklet sites, *p<0.05). (B) A greyscale image of wild-type (WT) TRPV4-transduced HEK293 cells, left; representative F/F0 traces from the WT and mutant TRPV4-transduced HEK293 cells, middle; and a bar graph comparing TRPV4 channel activity per site in the WT and mutant TRPV4-transduced HEK293 cells, right (n=25–48 sparklet sites, *p<0.05). (C) A comparison of burst open times for each quantal level between the control and proband fibroblasts (left) and WT and mutant TRPV4-transduced HEK293 cells (n=3–27 sparklet sites for fibroblasts; 10–24 sites for HEK293 cells; *p<0.05, right). The quantal levels were determined from the all-point histogram (see online supplementary figure S11).
We postulated that this novel gain-of-function mutation increases sparklet activity in proband fibroblasts and TRPV4-V829WfsX3 cells due to longer channel openings rather than an increase in channel number per site. In the measurement of TRPV4 Ca2+ sparklets, the majority of the TRPV4 sparklets lasted more than 1 s, indicating that each sparklet event may represent a burst of channel openings, as Loukin et al14 found single TRPV4 channel openings to last only a few milliseconds. To compare TRPV4 channel open times, fractional fluorescence (F/F0) traces were analysed to determine the mean burst open times at each quantal level, finding twofold longer mean burst open times for each quantal level in proband fibroblasts compared with control fibroblasts (figure 2C). Similarly, the mean burst open times were longer for all quantal levels in TRPV4-V829WfsX3 cells compared with TRPV4-WT cells (figure 2C). Thus, this mutation leads to a gain of function of TRPV4 channels by impeding channel closure.
Discussion
This study identifies a novel mutation in TRPV4 in a family affected with inherited osteonecrosis of the femoral head. The genetic finding is supported by identification of a TRPV4 gain of function in cultured cells. Strengths of the study include the genetic support provided by segregation of the mutation within the family and functional measurements performed on proband-derived fibroblasts and transduced HEK293 cells. Limitations of the study include the availability of a single family, which adds difficulty to eliminating all variant candidates in an autosomal-dominant model of inheritance and a lack of available phenotype-relevant cell types for functional measurements or an animal model to provide additional evidence of causation.
Previous studies investigating TRPV4 C-terminal truncations found loss of TRPV4 activity, contrary to a gain of function.13 ,15 As the frameshift deletion alters residues within a calmodulin-binding domain, altered TRPV4 channel regulation by calmodulin could be the molecular basis for the TRPV4 gain of function and will be investigated in addition to other TRPV4 regulatory factors and binding partners. Furthermore, SNPs in three glutamate receptor genes were recently identified as risk factors of glucocorticoid-induced osteonecrosis in a genome-wide association study.16 As Ca2+ influx is known to modulate release of chemical transmitters including glutamate in the brain, this finding could implicate TRPV4 in glucocorticoid-induced osteonecrosis.17
The pathogenesis of non-traumatic osteonecrosis is complex, involving compromised vasculature in the development of bone loss that is joint-specific. Interestingly, TRPV4 is known to regulate vascular tone as well as osteoclastic differentiation while also exhibiting mechanosensitive properties, proposed to play a role in the sensing of weight loading essential to bone development.6 ,18–20 The femoral head lacks collateral blood vessels, increasing risk for subchondral fracture and collapse. It is also a weight-bearing joint undergoing constant mechanical stress, which could promote localised TRPV4 activation in addition to systemic activation by the TRPV4 gain-of-function mutation. Together, this could cause Ca2+ overload, leading to endothelial dysfunction and vasoconstriction, resulting in bone loss at the hip. TRPV4-mediated vasoconstriction was recently demonstrated in the vascular endothelium by G protein-coupled receptor potentiation of TRPV4 through COX-dependent prostanoid production and thromboxane receptor activation.18 While TRPV4 is expressed in endothelium,6 ,7 ,18 measurements of TRPV4 function in human platelets have not been well described. However, in a washed human platelet aggregation assay, no response to GSK101 was found (R. Ramachandran, personal communication, 2015), suggesting that TRPV4 activation in endothelial cells is the most plausible origin of the microthrombotic process underlying osteonecrosis, possibly via a thromboxane-related mechanism. Our findings identify an association between TRPV4 and inherited osteonecrosis of the femoral head, providing additional insight into osteonecrosis pathogenesis and reinforcing the importance of TRPV4 in vascular endothelium and bone homeostasis.
Acknowledgments
The authors gratefully acknowledge the family members for volunteering their time for the clinical investigation and providing DNA samples; Dr Lambert Busque for providing DNA samples of the sporadic osteonecrosis cohort built in collaboration with EJH, CS and Dr Jeannine Kassis; David Hill-Eubanks, David Watkins and Michael Churchill-Smith for manuscript revision; Ildiko Horvath for help with illustrations; and Morley D Hollenberg and Rithwik Ramachandran for providing unpublished data and scientific discussion. We thank the Institute for Research in Immunology and Cancerology, Genomics Platform, Université de Montréal, for Bioanalyzer and qPCR measurements.
References
Footnotes
Contributors CS and DSR conceived the study. CS, EJH, DSR and WM designed the study. MTN and SKS designed functional assays and analysis. CS and WM coordinated the study. CS and EJH identified participants. CS, EJH, LR and CC evaluated and diagnosed participants. CS and TW isolated samples and collected data from participants. JM supervised exome sequencing and identified candidate genes. JCZ carried out exome sequencing. JCZ and MP analysed exome sequencing. DSR supervised genetic analysis. TW, WM, MP and BA carried out genetic analysis. WM generated cell lines, carried out immunological assays and wrote the initial manuscript. SKS and KH carried out functional assays and analysis. CS, WM, SKS and MTN wrote the manuscript and TW, MP and BA contributed to the manuscript. All authors reviewed the manuscript.
Funding This work was supported by the Montreal General Hospital Foundation and CS is supported by awards from the Canadian and US Leukemia & Lymphoma Societies (US LLS 6341-11), the Fonds de la Recherche du Québec-Santé (FRQS), and the Yvon Boulanger Foundation. EJH is supported by the Canadian Institutes of Health Research and the FRQS-sponsored Réseau de recherche en santé buccodentaire et osseuse. This work was also supported by NIH awards HL121484 to SKS, HL044455, 1P01HL095488, R37DK053832, R01HL098243 and R01HL121706 to MTN, Totman Medical Research Trust, and Fondation Leducq to MTN.
Competing interests None declared.
Ethics approval McGill University Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.