Article Text

Abstract
Background Ciliopathies are an extensive group of autosomal recessive or X-linked disorders with considerable genetic and clinical overlap, which collectively share multiple organ involvement and may result in lethal or viable phenotypes. In large numbers of cases the genetic defect remains yet to be determined. The aim of this study is to describe the mutational frequency and phenotypic spectrum of the CEP120 gene.
Methods Exome sequencing was performed in 145 patients with Joubert syndrome (JS), including 15 children with oral-facial-digital syndrome type VI (OFDVI) and 21 Meckel syndrome (MKS) fetuses. Moreover, exome sequencing was performed in one fetus with tectocerebellar dysraphia with occipital encephalocele (TCDOE), molar tooth sign and additional skeletal abnormalities. As a parallel study, 346 probands with a phenotype consistent with JS or related ciliopathies underwent next-generation sequencing-based targeted sequencing of 120 previously described and candidate ciliopathy genes.
Results We present six probands carrying nine distinct mutations (of which eight are novel) in the CEP120 gene, previously found mutated only in Jeune asphyxiating thoracic dystrophy (JATD). The CEP120-associated phenotype ranges from mild classical JS in four patients to more severe conditions in two fetuses, with overlapping features of distinct ciliopathies that include TCDOE, MKS, JATD and OFD syndromes. No obvious correlation is evident between the type or location of identified mutations and the ciliopathy phenotype.
Conclusion Our findings broaden the spectrum of phenotypes caused by CEP120 mutations that account for nearly 1% of patients with JS as well as for more complex ciliopathy phenotypes. The lack of clear genotype–phenotype correlation highlights the relevance of comprehensive genetic analyses in the diagnostics of ciliopathies.
- Clinical genetics
- Developmental
- Genetics
- Molecular genetics
- Neurosciences
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/
Statistics from Altmetric.com
Introduction
Ciliopathies are an extensive group of autosomal recessive or X-linked disorders caused by defects of the primary cilium, a highly conserved subcellular organelle found in most vertebrate cell types. Pathogenic mutations in genes required for proper processes involved at the primary cilium lead to disorders among which are cystic kidneys, retinal degeneration, intellectual disability, infertility and skeletal alterations. There is considerable genetic and clinical overlap among distinct ciliopathy syndromes that collectively share multiple organ involvement and may result in lethal or viable phenotypes.1
One of the most frequent ciliopathies is Joubert syndrome ((JS) MIM213300) that is uniquely characterised by a malformation of the cerebellum and brainstem known as the ‘molar tooth sign’ (MTS). This feature is defined by an abnormally deep interpeduncular fossa, elongated, thick and maloriented superior cerebellar peduncles and absent or hypoplastic cerebellar vermis, together giving the appearance of a ‘molar tooth’ on axial brain MRI through the junction of the midbrain and hindbrain (isthmus region). Besides this pathognomonic feature, the clinical and neuroimaging phenotype of JS and related disorders is heterogeneous, with frequent involvement of retina, kidneys, liver and skeleton and variable occurrence of additional brain malformations such as polymicrogyria, corpus callosum dysgenesis, occipital encephalocele and tectal dysplasia.2–4 The association of tectal and cerebellar dysraphia with occipital encephalocele (TCDOE) was first described by Friede in 1978.5 Only a handful of cases with TCDOE have been reported so far in the literature and the underlying genetic mechanism remains undetermined. Based on conventional neuroimaging and diffusion tensor imaging findings, TCDOE was recently suggested to be part of the JS spectrum.6
The severe end of the range of ciliopathies is represented by Meckel syndrome ((MKS), MIM249000), a lethal disorder characterised by cystic dysplastic kidneys, bile duct proliferation of the liver, occipital encephalocele, postaxial polydactyly, pulmonary hypoplasia due to oligohydramnios and occasional skeletal involvement such as bowing of long bones.7 Of the 12 genes identified as causative of MKS, 9 are also associated with JS.8 In addition, several families with occurrence of JS and MKS in the same sibship have been reported,9 ,10 which confirms that MKS and JS are allelic disorders.
Despite the impressive acceleration in gene discovery provided in recent years by the advent of exome sequencing, it is estimated that the currently known ciliopathy genes are responsible only for approximately 55%–60% of JS and up to 70% of patients with MKS,2 ,11 while a genetic diagnosis is not reached in the remaining individuals.
Recently, mutations in CEP120 were reported in four individuals diagnosed with Jeune asphyxiating thoracic dystrophy (JATD), a skeletal ciliopathy.12 Here we report the identification of mutations in the CEP120 gene in a spectrum of phenotypes ranging from mild classical JS to more severe conditions with overlapping features of distinct ciliopathies that include MKS, JATD and oral-facial-digital (OFD) syndromes. In addition, we propose CEP120 mutations as a genetic cause of TCDOE.
Patients and methods
Patients
Two large cohorts of patients with a diagnosis of JS or MKS were selected at The Rockefeller University (New York, USA) and the CSS-Mendel Institute (Rome, Italy). The unique inclusion criterion for JS was the presence of the MTS on neuroimaging; MKS was diagnosed in presence of at least two of the classical signs (occipital encephalocele, renal cystic dysplasia, postaxial polydactyly) and a normal karyotype. Detailed clinical data were obtained from the referring clinicians who filled in a standardised questionnaire. Neuroimaging studies were qualitatively reviewed for the presence of supratentorial and infratentorial morphological brain anomalies. All families provided written informed consent, according to institutional guidelines. Ethics approval has been obtained by the Ethics Committees of the Rockefeller University, the IRCCS Casa Sollievo della Sofferenza Institute and the UZ Leuven.
Exome sequencing
Exome sequencing was performed in 145 patients with JS, including 15 children with OFD syndrome type VI (OFDVI), and in 21 MKS fetuses. Moreover, exome sequencing was performed in one fetus with TCDOE, the MTS and additional skeletal abnormalities.
In solution, exome capture was carried out using the SureSelect Human All Exome 50 Mb Kit (Agilent Technologies, Santa Clara, California, USA) with 150 bp paired-end read sequences generated on a HiSeq2000 (Illumina, San Diego, California, USA). Sequences were aligned to hg19 and variants identified through the Genome Analysis Toolkit (GATK) pipeline.13 Variations were annotated with in-house software and the SeattleSeq server.14 Identified variants were checked against public databases dnSNP146, Exome Variant Server and ExAC. Currently known JS, MKS and OFD genes were specifically analysed for the presence of pathogenic mutations.
Target resequencing of 120 ciliary genes
In order to assess the mutational frequency and phenotypic spectrum of CEP120 mutations, this gene was included in a panel of 120 previously described and candidate ciliopathy genes (among which all known JS, MKS and OFD genes) used for next-generation sequencing-based targeted sequencing in a cohort of 346 probands with a phenotype consistent with JS or related ciliopathies. Targeted resequencing was performed on a Solid 5500xL platform using the TargetSeq Custom Enrichment System (Thermo Fisher Scientific), according to manufacturers' protocols. Briefly, DNA libraries were fragmented and coupled with specific adaptors, enriched for the set of genes of interest, barcoded for pooling and then amplified by emulsion PCR, prior to high-throughput sequencing. Variants were detected by means of the HaplotypeCaller software package of the GATK suite and filtered so that to include only variants covered by at least 20 reads and with mapping quality values exceeding a Phred-score of 30. Variants reported as validated polymorphisms with minor allele frequency (MAF) of ≥0.01 in publicly available human variation resources (dbSNP146, 1000 Genomes, National Heart, Lung, and Blood Institute Exome Sequencing Project Exome Variant Server (EVS)) as well as variants present in in-house control individuals with MAF of ≥0.01 were filtered out.
Validation of CEP120 variants
All identified CEP120 variants were verified by bidirectional Sanger sequencing in the proband as well as available family members, to check segregation with the disease and carrier status of the respective parents. Potential pathogenicity was predicted using the software tools SIFT, PolyPhen-2, PROVEAN and Mutation Assessor. Conservation of affected residues was assessed by the Clustal Omega software. Reference sequences applied were CEP120 gene, NM_153223.3, CEP120 protein and NP_694955.2. All identified variants have been submitted to the Leiden Open (source) Variation Database (LOVD) gene variant database.
Websites for all software and databases are listed in online supplementary resources section.
Supplementary resources section
Results
Identification of CEP120 mutations
Biallelic mutations in the CEP120 gene were identified in four probands with JS and two fetuses with overlapping ciliopathy phenotypes, all segregating with the disease within the families (figure 1A, B; three variants were present in homozygous and six in compound heterozygous state). Overall, nine distinct mutations were detected in this study, two of which were predicted to result in a truncated protein (one nonsense mutation and one 1 bp deletion). One of the variants comprised a splice-site mutation and the remaining six were missense mutations affecting highly conserved residues, all consistently predicted to be deleterious by four distinct in silico predictors (see figure 1C and online supplementary table S1). One of the missense mutations identified in this study (p.Ala199Pro) was previously reported by Shaheen and collaborators.12 Seven out of nine mutations were absent from the public reference databases (dbSNP, EVS and ExAC), while two of them (p.Ala199Pro and p.Leu712Phe) were found in dbSNP (rs114280473 and rs367600930, respectively), ExAC or EVS, however, at very low frequency (MAF<0.5%) and never in a homozygous state. All identified mutations were absent from our combined in-house database.

Pedigrees and schematic representation of CEP120. (A) Genomic structure and mRNA transcript of CEP120 full-length isoform with 21 exons is shown. Untranslated regions (UTR's) are represented by half-height boxes. The location of the mutations is indicated by M1–M9. (B) Pedigrees of the affected families with ancestries of Italy (COR391), USA (MTI-143), Palestine (MTI-991), India (MTI-1516), Turkey (Meckel syndrome (MKS)-2930) and Belgium (SW-476410), respectively, is shown demonstrating the segregation of the compound heterozygous mutations in non-consanguineous families and homozygous mutations in consanguineous families. (C) Evolutionary conservation of affected amino acid residues in CEP120 is shown. The mutated amino acids are indicated in grey and completely conserved in all species shown. JS, Joubert syndrome; M, mutation; TCDOE, tectocerebellar dysraphia with occipital encephalocele. See online supplementary figure S1 for the chromatograms of mutation M1 in CEP120 (remaining mutation data not shown).
Supplementary table
Supplementary figure
Interestingly, one of the mutations consisted of a homozygous intronic deletion leading to intron retention in family MTI-991. Located at 5 bp from the splice donor site of exon 2, a 6 bp deletion led to the use of an alternative splice site 483 bp downstream (c.49+5_49+10delins477), in approximately 5% of mRNA, which was predicted to result in a truncated protein after 14 amino acids (p.Gly+1Aspfs*14) (see online supplementary figure S1). The remaining mRNA product showed normal splicing from exon 2 to exon 3. Mutations in the known genes causative of JS and other ciliopathies were excluded in all patients with CEP120 mutations.
Clinical features of mutated subjects
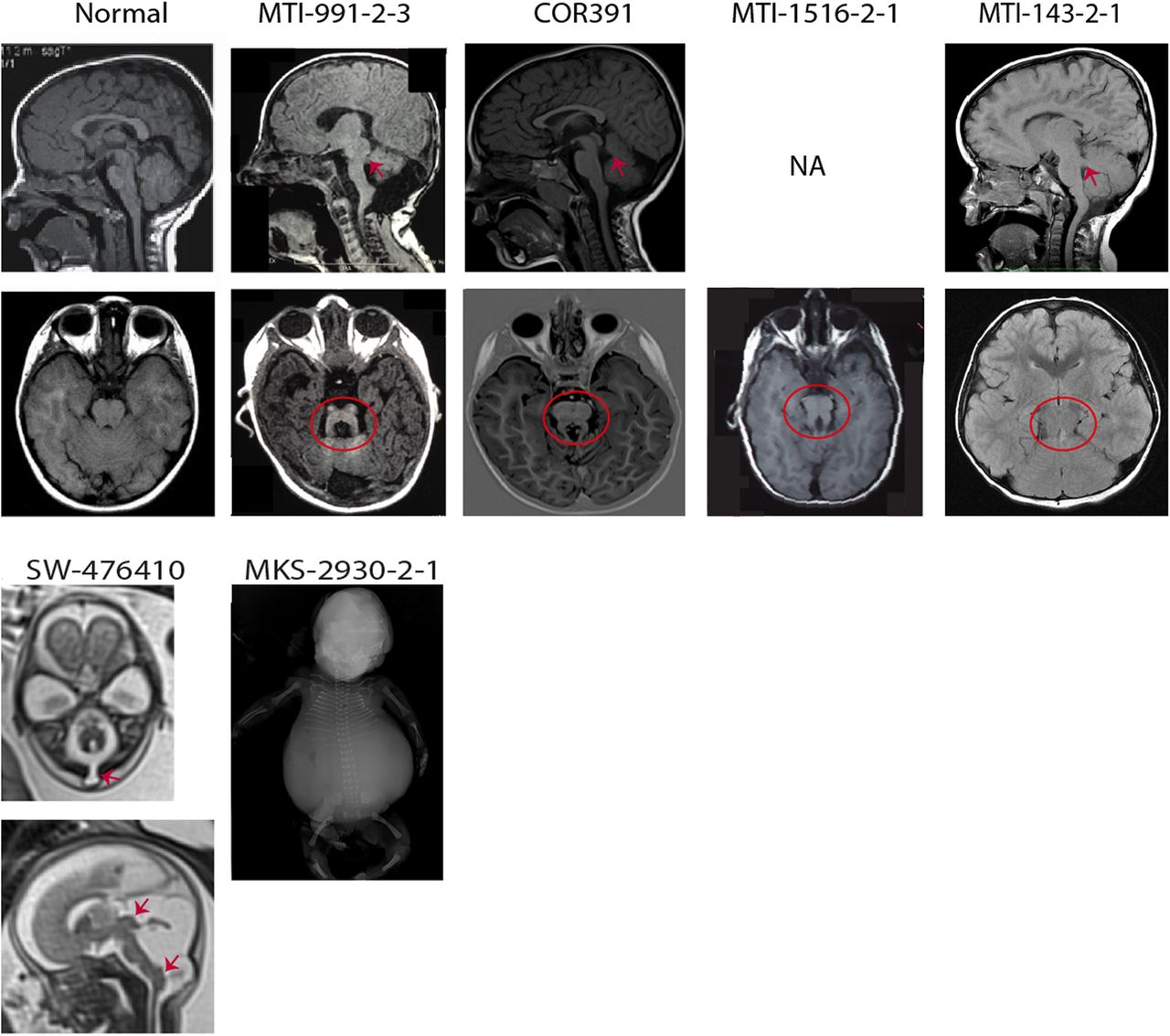
The clinical and neuroimaging features of the four probands with JS carrying CEP120 mutations are summarised in table 1 and figure 2. They all presented with a neurological phenotype consisting of hypotonia, developmental delay and cognitive impairment. Ataxia and neonatal breathing abnormalities were reported in two individuals (MTI-143, MTI-1516), while abnormal ocular movements were observed in a single family. There was no involvement of other organs such as the retina, kidneys, liver and skeleton.
Clinical, neuroimaging and genetic features of children with JS having CEP120 mutations
{kind=link}
{kind=link}
Neuroimaging or neuropathological findings of probands with CEP120 mutations are shown. Upper panel: Sagittal T1-weighted and axial T1-weighted/coronal fluid attenuation inversion recovery MRIs of a healthy subject and patients with Joubert syndrome (JS) having CEP120 mutations show thickened and maloriented superior cerebellar peduncle (upper arrows in MTI-991-2-3 and MTI-143-2-1), rostral shifting of the fastigium of the fourth ventricle (upper arrow in COR391), deepened interpeduncular fossa and constituting the ‘molar tooth sign’ (circled). Lower panel: SW-476410: axial and midsagittal T2-weighted fetal MRIs at 23 weeks of gestation show a suboccipital encephalocele (arrow on the axial image), severe hypoplasia of the cerebellar vermis and tectal dysplasia (upper arrow on the sagittal image) consistent with the diagnosis of tectocerebellar dysraphia with occipital encephalocele (TCDOE). In addition, enlargement of the posterior fossa and dorsal protuberance of the lower brainstem (lower arrow on the sagittal image) are noted; Meckel syndrome (MKS)-2930-2-1: a fetogram after termination of pregnancy in the second trimester reveals a marked abdominal distention, a narrow bell-shaped thorax with short ribs, rhizomelic limb shortening and bowing of long bones.
Furthermore, mutations in CEP120 were detected in two fetuses (table 2). The first one (MKS-2930) received the diagnosis of MKS in the second trimester of pregnancy, due to the presence of cystic dysplastic kidneys, occipital encephalocele and polydactyly. In addition, postmortem examination after pregnancy termination disclosed a narrow bell-shaped thorax with short ribs, rhizomelic limb shortening, bowing of long bones, microphthalmia, oral-facial defects (lobulated tongue with multiple frenula and cleft soft palate), ambiguous genitalia and anal atresia. Data on liver involvement were not available.
Pathological and genetic features of fetuses with CEP120 mutations
The second fetus (SW-476410) was diagnosed in utero with TCDOE in the spectrum of JS, due to the presence of a suboccipital encephalocele, dysplastic tectum, severe hypoplasia of the cerebellar vermis and the MTS, as shown by fetal MRI at 23 weeks of gestation. In addition, postmortem examination revealed cleft palate, narrow thorax with short ribs and secondary lung hypoplasia and discrete rhizomelic shortening of limbs. Liver and kidneys were normal and no polydactyly was observed.
Discussion
We report for the first time the occurrence of CEP120 mutations in 4 out of 491 patients with JS (0.8%). Of note, all of them presented with a mild, purely neurological phenotype, adding CEP120 to the growing list of ciliary genes that are associated with this relatively benign phenotype.3
Furthermore, we identified CEP120 mutations in two fetuses (MKS-2930 and SW-476410) with more complex phenotypes and overlapping features of distinct ciliopathies. Both showed central nervous system malformations characterised by occipital/suboccipital encephalocele and enlarged posterior fossa. In addition, one fetus (SW-476410) showed marked hypoplasia of the cerebellar vermis, dysplasia of the tectal plate and a posterior protuberance of the lower brainstem.
The association of hypoplasia or agenesis of the cerebellar vermis, tectal malformation (reminiscent of the tectal beaking characteristic of the Chiari type 2 malformation) and occipital encephalocele was first described by Padget and Lindburg in 1972.15 In 1978, Friede introduced the term ‘tectocerebellar dysraphia with occipital encephalocele’ (TCDOE) to describe this constellation of findings.5 To our knowledge, in literature this entity has only been described as a clinical diagnosis in eight cases.6 ,16–21 Neuroimaging studies showed elongated and horizontally orientated superior cerebellar peduncles reminiscent of the MTS in all reported patients and a deepened interpeduncular fossa in three of them. In addition, diffusion tensor imaging in one child revealed absence of the decussation of the cerebellar peduncles, as previously shown in patients with JS.4–6 In the fetus SW-476410, the suboccipital encephalocele was located at the level of the foramen magnum. This is an unusual location for encephalocele and has previously been reported only in JS.4 ,22 Furthermore, in another published case, TCDOE was associated with a congenital heart disease and situs inversus, reinforcing the hypothesis of an underlying ciliopathy.21 However, no causative genes for the TCDOE phenotype had been found to date and, to our knowledge, this is the first report of mutations in a ciliary gene being causative of this exceptional condition. The finding of mutations in CEP120 in one fetus with TCDOE further supports the observation that TCDOE represents a morphological phenotype within the spectrum of JS.
Interestingly, both CEP120-mutated fetuses showed a small, bell-shaped thorax with short ribs and rhizomelic shortening of limbs, which are typical features of the skeletal ciliopathy known as JATD. Besides these malformations, the typical JATD phenotype is characterised by polydactyly, cystic dysplastic kidneys and bile duct proliferation of the liver, all features that are also found within the spectrum of MKS.23 However, only one of the fetuses presented with postaxial and preaxial polydactyly and cystic kidneys and therefore was initially diagnosed with MKS.
The co-occurrence of JS and the core signs of JATD (mainly a small thorax with secondary respiratory distress), was previously described in some living patients.23 In addition, mutations in two genes, CSPP1 and KIAA0586, were reported to cause both JS and JATD, either isolated or in variable combination.24–31 Recently, a founder missense mutation of CEP120 (p.Ala199Pro) was identified in four Saudi Arabian families presenting the full-blown JATD phenotype, associated with cerebellar vermis hypoplasia in two patients and MTS in one of them.12 Remarkably, despite his Flemish origin without Arabian ancestors, one fetus in the present study (SW-476410) was found to carry the same founder mutation in combination with a nonsense mutation, leading to the hypothesis that this particular mutation might predispose to the development of skeletal defects.
Noteworthy, the two fetuses with JATD features in our study also showed oral-facial defects including cleft lip, tongue hamartomas and bifid or lobulated tongue. These oral-facial findings were also reported in three of four CEP120-mutated patients with JATD reported previously, suggesting that they may be commonly associated with the skeletal phenotype and specific to CEP120 mutations in both lethal or surviving cases. The co-occurrence of features, each of which typical of distinct ciliopathies (short rib polydactyly, JS/MKS spectrum and OFD syndromes) in the same patient is an intriguing finding that has already been reported in the literature. For instance, Thomas and collaborators identified mutations in the TCTN3 gene in individuals with classical JS and in fetuses with Mohr–Majewski syndrome (also called OFD IV): an extremely severe condition characterised by oral-facial features, polydactyly, encephalocele and MTS, as well as renal, hepatic and skeletal anomalies.32 Moreover, patients with JS can also present oral-facial defects and polydactyly (mainly preaxial and mesoaxial), a rare association termed OFDVI.33 So far, mutations in TMEM216, OFD1 and C5Orf42 were found mutated in a minority of patients with OFDVI.9 ,34–37 In the present study, CEP120 was sequenced in a cohort of 15 patients with OFDVI negative for mutations in currently known JS genes, but no pathogenic mutations were identified. This result suggests that CEP120 may have little contribution to this specific JS phenotype.
Cep120 is a centrosomal protein that is essential for microtubule organisation and centriolar assembly and elongation.38 ,39 In mouse fetal brain, Cep120 is expressed in neural progenitors during neocortical development and plays an important role in neuronal migration and maintenance of the neural progenitor pool.40 Its downregulation alters microtubule organisation and centrosome motility, with consequent defective axonal growth and impaired neuronal migration.41 Accordingly, the conditional ablation of Cep120 in the mouse central nervous system led to aberrant centriolar duplication, failed ciliogenesis and altered Sonic Hedgehog pathway activity in the granule neuron progenitors, resulting in hydrocephalus and severe cerebellar hypoplasia.42 In addition, splice-blocking morpholino injected zebrafish knocking down Cep120 presented with a range of phenotypes including a centrally curved tail, hydrocephalus, otolith defects and craniofacial defects.12
Of note, Cep120 was found to interact with Talpid3,42 the homologue of human KIAA0586, in which mutations give rise to a similar spectrum of ciliopathies, ranging from a mild, pure JS to a more complex phenotype with overlapping features of JS and JATD.27–31 This increasing range of phenotypes associated with mutations in one and the same gene argues for the need of broad next-generation sequencing-based approaches, to allow the detection of novel, rare phenotypes associated with ciliary gene mutations.
The mechanism through which mutations in the same gene may cause such wide phenotypic variability remains unexplained. Genotype–phenotype correlations have been proposed for ciliopathy genes such as TMEM67, in which two loss-of-function mutations were found to cause MKS while the presence of at least one hypomorphic mutation resulted in non-lethal phenotypes.43 However, this is not obvious for CEP120, as the MKS fetus (MKS-2930) was homozygous for a missense mutation, while at least one JS patient (MTI-991) carried a splice-site mutation that was shown to introduce a premature stop codon in part of the product. Growing evidence is demonstrating that ciliopathies might undergo an oligogenic mode of inheritance and that heterozygous mutations in distinct genes can modulate the clinical expression of the main recessive mutations. For instance, the common variant p.Arg830Trp in the AHI1 gene was found to significantly enhance the frequency of retinal disease in patients with NPHP1-associated nephronophthisis.44 The identification of such genetic modifiers, as well as of other factors able to influence the penetrance and expression of ciliary gene mutations, represents one of the biggest challenges of current research on ciliopathies, with significant implications for diagnosis, management and counselling of patients and their families.
In conclusion, this study adds CEP120 to the growing list of causative genes for a variety of clinically distinct but overlapping ciliopathies. In addition, we provide the primary molecular diagnosis in a case of TCDOE and confirm the classification of this morphological phenotype within the spectrum of JS.
Acknowledgments
The authors thank the affected children and their families for their invaluable contributions to this study. They also thank the Broad Institute (U54HG003067 to E. Lander), the Yale Center for Mendelian Disorders (U54HG006504 to R. Lifton and M. Gunel) for sequencing support.
References
Footnotes
SR and MR contributed equally.
Contributors SR and MR: substantial contributions to the conception or design of the work; the acquisition, analysis or interpretation of data for the work (performed and analysed experiments). MI, ROR, AM, DM and TM: substantial contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work (CEP120 sequencing and validation). LA, UA, EB, SD'A, BDK, HK, SB, IK, PRM, TMK, JVK, PM, AP and RP: substantial contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work (clinical characterisation of study participants). HVE, JGG and EMV: substantial contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work (collection and clinical characterisation of study participants, exome studies, securing funding). In addition, all of the authors drafted the work or revised it critically for important intellectual content and gave final approval of the version to be published; they agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding This work was supported the European Research Council (ERC Starting Grant 260888 to EMV), Telethon Foundation Italy (GGP13146 to EMV), NI H grants (R01NS041537, R01NS048453, R01NS052455, P01HD070494, P30NS047101 to JGG); the Howard Hughes Medical Institute and Simons Foundation (to JGG) and Concerted Research Actions KULeuven GOA/12/015 and funding from the Belgian Science Policy Office Interuniversity Attraction Poles programme (IAP P7/43-BeMGI to HVE). HVE is a clinical investigator of FWO-Vlaanderen.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Ethics approval has been obtained by the Ethics Committees of the Rockefeller University, the IRCCS Casa Sollievo della Sofferenza Institute and the UZ Leuven.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Additional unpublished sequences are available upon request at the corresponding author.