Article Text

Abstract
Background Auditory neuropathy spectrum disorder (ANSD) is a form of hearing loss in which auditory signal transmission from the inner ear to the auditory nerve and brain stem is distorted, giving rise to speech perception difficulties beyond that expected for the observed degree of hearing loss. For many cases of ANSD, the underlying molecular pathology and the site of lesion remain unclear. The X-linked form of the condition, AUNX1, has been mapped to Xq23-q27.3, although the causative gene has yet to be identified.
Methods We performed whole-exome sequencing on DNA samples from the AUNX1 family and another small phenotypically similar but unrelated ANSD family.
Results We identified two missense mutations in AIFM1 in these families: c.1352G>A (p.R451Q) in the AUNX1 family and c.1030C>T (p.L344F) in the second ANSD family. Mutation screening in a large cohort of 3 additional unrelated families and 93 sporadic cases with ANSD identified 9 more missense mutations in AIFM1. Bioinformatics analysis and expression studies support this gene as being causative of ANSD.
Conclusions Variants in AIFM1 gene are a common cause of familial and sporadic ANSD and provide insight into the expanded spectrum of AIFM1-associated diseases. The finding of cochlear nerve hypoplasia in some patients was AIFM1-related ANSD implies that MRI may be of value in localising the site of lesion and suggests that cochlea implantation in these patients may have limited success.
- Clinical genetics
- Genetic heterogeneity
- Mutation
- Auditory neuropathy spectrum disorder
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/
Statistics from Altmetric.com
Introduction
Auditory neuropathy spectrum disorder (ANSD) is characterised by absent or severely abnormal inner hair cell (IHC) function as measured by auditory brainstem responses (ABRs), with preservation of outer hair cell (OHC) function as indicated by otoacoustic emission (OAE) and/or cochlear microphonic (CM) testing. First described by Starr et al in 1996,1 patients with ANSD present with variable degrees of unilateral or bilateral hearing impairment accompanied by poor speech discrimination and poor word understanding especially in the presence of noise. The prevalence of ANSD varies from 0.5% to 15% among hearing-impaired patients, with an incidence of about 13% in children with severe-to-profound hearing loss.2–4 Consistent with physiological tests of auditory function, ANSD can be caused by lesions of the IHC, IHC–auditory nerve synapse, auditory nerve or auditory cortex.5–7
In many cases of ANSD, the molecular pathology remains unclear, with underlying aetiologies running the gamut of genetic abnormalities, toxic/metabolic derangements, infections, immunological causes and drugs.8 ,9 Forty per cent of ANSD is estimated to have a genetic basis with autosomal-dominant, autosomal-recessive, mitochondrial and X-linked inheritance all reported.3 The list of causative genes includes OTOF, PJVK, DIAPH3 and mtDNA (m.1095T>C) in non-syndromic ANSD and PMP22, MPZ, TMEM126A and DDDP in syndromic ANSD, although other genetic aetiologies await discovery.2
In 2006, we reported a large Chinese family with X-linked progressive auditory and peripheral sensory neuropathy, and mapped this ANSD locus (AUNX1) to chrXq23-27.3.10 Using whole-exome sequencing (WES), we have identified the causal AUNX1 gene as AIFM1 and show that variants in this gene are a common cause of familial and sporadic ANSD. This finding is noteworthy because AIFM1 mutations are also associated with mitochondrial encephalomyopathy, prenatal ventriculomegaly and Cowchock syndrome, three disorders characterised by developmental disabilities, motor dysfunction, muscle weakness and brain abnormalities as resolved by MRI.11–13 Our work expands the spectrum of AIFM1-associated phenotypes and mandates screening of AIFM1 in small pedigrees with apparent autosomal-recessive ANSD if X-linked inheritance cannot be excluded.
Methods
Family ascertainment and clinical evaluation
Five unrelated Chinese families (AUNX1, 7170, 0223, 2724 and 2423) and 93 sporadic male patients diagnosed with ANSD were ascertained through the Department of Otolaryngology, Head and Neck Surgery, Chinese PLA General Hospital, from November 2000 to June 2014. The phenotype of the AUNX1 family has been reported and includes auditory neuropathy and delayed peripheral sensory neuropathy inherited in an X-linked recessive pattern. The other four families had a similar clinical phenotype. In all cases, genes commonly involved in ANSD such as OTOF, PJVK and DIAPH3 were excluded by Sanger sequencing. Acquired causes of ANSD such as prematurity, hyperbilirubinemia, anoxia, hypoxia, congenital brain abnormalities, perinatal intracranial haemorrhage, asphyxia and ototoxic drug exposure were excluded by medical history.
All participants were examined by a multidisciplinary team of healthcare providers that included a neurologist, otolaryngologist and audiologist. The evaluation consisted of a comprehensive medical history, physical examination with careful clinical assessment for peripheral neuropathies, pure-tone audiometry, tympanometry, acoustic reflex testing, ABR, distortion-product OAE testing and electrocochleography. The diagnosis of ANSD was based on recognised criteria (Guidelines Development Conference at NHS 2008, Como, Italy). Neurological examinations included assessments of cranial nerve function, motor activity, muscle weakness, sensory impairment and reflexes. Electrophysiological studies including needle electromyography (EMG), nerve conduction velocity (NCV) and somatosensory evoked potential (SEP) in seven patients from the four unrelated families were carried out. The EMG was performed in the abductor pollicis brevis, tibialis anterior and vastus lateralis muscles. Motor nerve conduction velocities (MCVs) of tibial, peroneal, median and ulnar nerves, as well as sensory nerve conduction velocities (SCVs) of sural, median and ulnar nerves, were obtained. Amplitudes of compound muscle action potential (CMAPs) and sensory nerve action potential (SNAPs) were measured from positive to negative peak values. The Mini Mental State Examination (MMSE) on the three patients (III: 1, III: 9 and III: 11) of family 2423 was conducted to assess the cognitive function. Select patients underwent MRI of brain, temporal high-resolution CT and electrocardiography. Serum enzymes related to energy metabolism, such as lactate dehydrogenase and creatine kinase, were measured.
Peripheral blood samples were obtained and genomic DNA was extracted according to standard procedures. WES was performed on one person from the AUNX1 family (III: 12) and three persons from the 0223 family (II: 1, III: 1 and III: 3). Results were confirmed and validated by Sanger sequencing in these persons and other available family members (figures 1B and 2A). Three other small families (7170, 2724 and 2423, figure 2B–D) and 93 unrelated sporadic cases with ANSD were analysed by Sanger sequencing. Five hundred ethnicity-matched individuals (250 men and 250 women) with normal hearing were recruited as normal controls.

Identification of the disease-causing AIFM1 mutation in the AUNX1 family segregating auditory and peripheral neuropathy. (A) Schematic genetic and physical map of the AUNX1 locus on chromosome Xq23-q27.3. The location of the AIFM1 gene is indicated (Mb, million bps). (B) The phenotype in AUNX1 family co-segregates with the c.1352G>A (p.R451Q) mutation in AIFM1. The genotypes at c.1352 for the family members are given: G or GG means hemi- or homozygous for the wild-type (WT) sequence, GA means c.1352G>A heterozygous, and A denotes the mutation in hemizygous form. Whole-exome sequencing was completed on subject III: 12. (C) Sequence chromatograms of exon 13 of AIFM1 show the c.1352G>A (p.R451Q) mutation (arrowhead) in affected males (hemizygote) and female carriers (heterozygote). A homozygous WT sequence is shown on the bottom. (D) Multiple sequence alignment depicts evolutionary conservation of amino acid residue Arg451 (red vertical bar) across human, bovine, mouse, chicken, Xenopus and zebrafish.

The four auditory neuropathy spectrum disorder families segregating AIFM1 mutations. Pedigree, sequence results and typical audiogram of each family are shown. Missense mutations c.1030C>T (p.L344F), c.778A>G (p.T260A), c.1264C>T (p.R422W) and c.1265G>A (p.R422Q) were identified in family 0223 (A), 7170 (B), 2724 (C) and 2423 (D), respectively. These mutations co-segregate with auditory and peripheral sensory neuropathy while carriers have normal hearing and sensory ability. The genotypes at c.1030, c.778, c.1264 and c.1265 for the available members in the corresponding family are given respectively. Whole-exome sequencing was completed on three persons in family 0223 (II: 1, III: 1 and III: 3). Needle electromyography and nerve conduction studies were performed on the individuals from family 7170 (IV: 2), family 0223 (III: 3), family 2724 (II: 4) and family 2423 (III: 1, III: 9 and III: 11). The Mini Mental State Examination was conducted on the three patients in family 2423 (III: 1, III: 9 and III: 11).
Whole-exome sequencing
Quantified, high-quality genomic DNA (2 µg per person) from one individual from family AUNX1 (III: 12) and three individuals from family 0223 (II: 1, III: 1 and III: 3) was used for WES. Each genomic DNA sample was captured using Agilent SureSelectXT Human All Exon V5 technology (Agilent Technologies, Santa Clara, California, USA) and enriched libraries were sequenced using the HiSeq 2000 platform (Illumina, San Diego, California, USA). Raw image files were processed on the Illumina Pipeline V.1.6 using default parameters, and sequences generated as 75–90 bp paired-end reads were aligned to NCBI37/hg19 assembly. Duplicate reads were removed using Picard (http://picard.sourceforge.net), and clean reads localised to the target region were collected and analysed by SOAPsnp (V.1.03).14 Local realignment of insertions and deletions (indels) and variant annotation were performed using the Genome Analysis Toolkit (http://www.broadinstitute.org/gatk/).15 By previously described criteria,16 ,17 the low-quality variations were filtered out.
Target enrichment was analysed using NGSrich.18 Variants were filtered against 1000 Genomes data, and all variants with a minor allele frequency (MAF) >1% were removed from the analysis. Functional annotation of genetic variants was performed using ANNOVAR (http://www.openbioinformatics.org/annovar/). Candidate variants were Sanger validated.
Mutation screening of AIFM1
AIFM1 (NC_000023.10) contains 16 exons. Thirty-two primers (16 primer pairs) were designed using Primer V.3.0 software and synthesised by Invitrogen by Life Technology (Beijing, China) to amplify each exon and exon–intron boundaries (see online supplementary table S1). PCR was performed with PE9700 thermocyclers (Applied Biosystems) using standard conditions. Amplified products from all ANSD cases and controls were gel purified and sequenced (ABI 3730, Applied Biosystems). Nucleotide alterations were identified by sequence alignment with the NCBI Reference Sequence (RefSeq) using DNAStar software V.5.0 (DNASTAR, Madison, Wisconsin, USA).
Evaluation of the pathogenicity
Pathogenicity was assessed using PolyPhen-2 (Polymorphism Phenotyping V.2, http://genetics.bwh.harvard.edu/pph2/index.shtml), SIFT (http://sift.jcvi.org/), Protein Variation Effect Analyzer (PROVEAN) (http://provean.jcvi.org/index.php) and Mutation accessor (http://mutationassessor.org/).
Immunofluorescent staining in mouse inner ear
All experimental procedures were approved by the Institutional Animal Care and Use Committee of the University at Buffalo that conform to the guidelines issued by the National Institutes of Health. Adult mice (C57, 2 months of age) were used for immunofluorescence studies. The cochlear tissues of basilar membrane, spiral ligament and the vestibular end-organ of saccule macula were micro-dissected out as has been described.19 ,20 After incubation at 4°C for 24 h with 1% Triton X100 and 5% goat serum in 0.1 M phosphate buffered saline (PBS) containing AIF primary antibody (rabbit monoclonal antibody against AIF, 1:100, Cat# ab32516, Abcam), the specimens were then incubated with tetramethylrhodamine isothiocyanate dextran-conjugated goat antirabbit secondary antibody (1:500, Cat# F6005, Sigma) in PBS for 2 h at room temperature. The speciments were next immersed in Alexa-488-conjugated phalloindin (1:200, Cat#A12379, Invitrogen) for 40 min to label the stereocilia and cuticular plate of the cochlear and vestibular sensory hair cells, and the F-actin in the gap-junction of marginal cells of stria vascularis. The nuclei of tissues were also labelled with 4′,6-diamidino-2-phenylindole dihydrochloride for 30 min. Immunoreactive products were observed under a confocal laser scanning microscope. As a negative control, the primary antibodies were omitted.
Results
Identification of missense mutations in AIFM1 by WES
To identify the causative variant at the AUNX1 locus, we completed exome sequencing of the proband in family AUNX1. Ninety-nine variants were identified within the AUNX1 locus. After filtering out synonymous changes and variants found in <85% of reads (inconsistent with X-linked inheritance in individual III: 12, an affected man), 24 variants remained: 17 non-synonymous variants, 2 in-frame indels and 5 frameshift indels. Based on incidence data for non-syndromic hearing loss and auditory neuropathy, we excluded variants with an MAF >0.001.21 ,22 Three variants remained—AIFM1 chrX 129267384:G>A; HS6ST2 chrX 131762528:G>A and VCX3A chrX 6452043:C>A—none of which are reported in the NHLBI Go Exome Sequencing Project (ESP) (6503 individuals) or the 1000 Genomes Project (1000G) (1092 individuals). Because VCX3A is expressed only in male germ cells, it was not considered further.23 ,24 Both of the remaining variants were confirmed in the proband by Sanger sequencing, but only AIFM1 p.R451Q co-segregated with the phenotype in the extended AUNX1 family (eight informative meioses were tested; figure 1A–C). This variant was not found in a screen of 500 normal-hearing ethnicity-matched controls (250 women, 250 men, 750 X chromosomes).
To identify the causative variant in the second family (0223), we completed exome sequencing of the proband (III: 1), his affected brother (III: 3) and unaffected father (II: 1). WES generated an average of 12.3 Gb of sequence, with at least 120× average coverage for each individual as paired-end, 90 bp reads, indicating the high quality of sequencing (see online supplementary figures S1 and S2 and table S2). After mapping to the reference genome sequence, >99.0% of the targeted bases were covered sufficiently to pass quality assessment for calling single-nucleotide polymorphisms (SNPs) and short indels (see online supplementary table S2). We identified an average of ∼20 700 SNPs in coding regions (exonic), an average of 129 variants (SNPs and indels) within 2 bp of an exon/intron boundary that may affect splicing, and an average of 1270 indels in coding regions (see online supplementary table S3). Because the two affected individuals share the causal variant when compared with their normal-hearing father, a total of 807 variants were retained after filtering against SNP and Indel databases including dbSNP 141, 1000G, Hapmap 8 and YH (see online supplementary tables S4 and S5). Among them, 213 variants (including 129 non-synonymous SNPs and splice sites, as well as 84 indels) were predicted to have a functional impact (see online supplementary tables S6 and S7). Because inheritance was consistent with an autosomal or X-linked recessive pattern based on family pedigrees, candidate pathogenic variants selected for further analysis were rare homozygous or hemizygous nonsense, missense, splice site and indel variants with allele frequencies of ≤0.005 in public variant databases. Combined with the predicted effect on protein function by SIFT, PolyPhen2 and Mutation Assessor programs, we identified five variants (including one rare SNP and four indels) to be candidates (see online supplementary table S8). After Sanger sequencing and genotyping in all available family members, the only variant segregating with the phenotype (auditory and peripheral sensory neuropathy) of family 0223 was c.1030C>T (p.L344F) in the AIFM1 gene (figure 2A). This variant is recorded in dbSNP (rs184474885, http://www.ncbi.nlm.nih.gov/projects/SNP) with a very low MAF (A=0.0005, 2/3775). Interestingly, although this variant is recorded as an SNP in 1000G, it was not found in the screen of 500 normal-hearing ethnicity-matched controls in Chinese populations (mentioned above).
Mutation spectrum of AIFM1 in familial and sporadic ANSD
To investigate the contribution of AIFM1 to ANSD in China, we screened this gene for mutations in our extended familial and sporadic ANSD cohort; identifying 10 more novel missense mutations in three additional families and 11 of 93 (10%) men with an ANSD phenotype (figure 3, table 1 and online supplementary table S9). In all cases of familial ANSD, the identified missense mutations (c.778A>G [p.T260A], c.1030C>T [p.L344F], c.1264C>T [p.R422W], c.1265G>A [p.R422Q], c.1352G>A [p.R451Q]) completely segregated with the auditory and peripheral sensory neuropathy phenotype, with female carriers not reporting any signs of ANSD or peripheral sensory neuropathy (figures 1B and 2). Two variants identified in familial ANSD, c.1030C>T (p.L344F) and c.1264C>T (p.R422W), were also detected in sporadic ANSD cases, with similar phenotypes (table 1). All of the 10 additional AIFM1 mutations were also absent in the screen of normal-hearing ethnicity-matched controls (750 X chromosomes). None of these variants were found in ESP or 1000G except c.1030C>T (p.L344F) (see online supplementary table S10).
Summary of the clinical phenotypes for cases with AIFM1 mutations

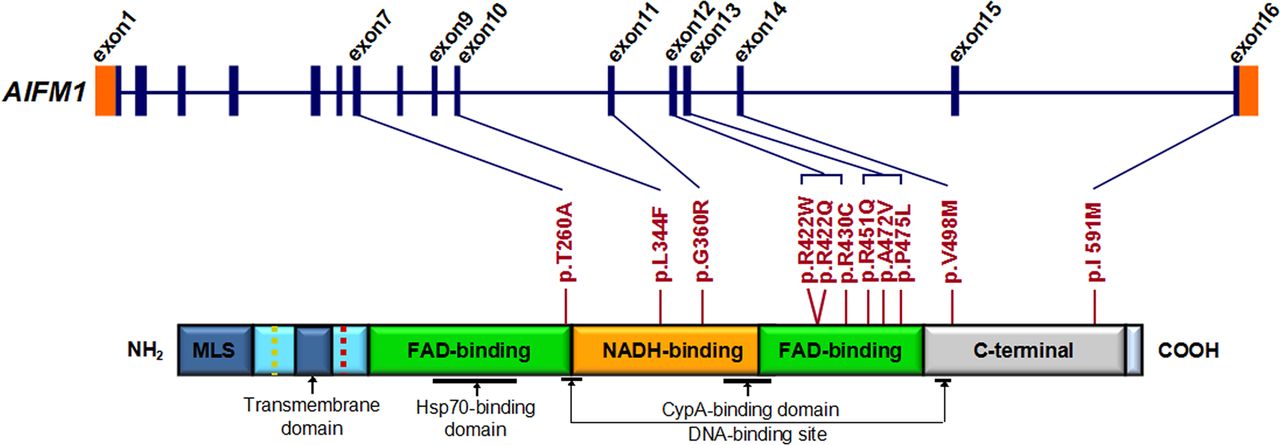
AIFM1 mutation screening in patients with familial and sporadic auditory neuropathy spectrum disorder (ANSD) with or without peripheral neuropathy. Graphical representation of AIFM1 structure (upper panel) and its encoded protein (lower panel). AIFM1 gene has 16 exons. As a flavoprotein with an oxidoreductase enzymatic activity, AIFM1 contains a flavin adenine dinucleotide (FAD)-bipartite domain (in green), a reduced nicotinamide adenine dinucleotide (NADH)-binding motif (in orange) and a C-terminal domain (in grey). It also has a mitochondria localisation sequence (in blue) located in the N-terminal region (reference: http://atlasgeneticsoncology.org//Genes/AIFM1ID44053chXq25.html). The positions of 11 mutations identified in familial and sporadic ANSD cases are shown between the two diagrams (blue lines and red bars). The c.1352G>A (p.R451Q) mutation identified in the AUNX1 family is located in exon 13, which corresponds to the second FAD domain. The longest isoform of AIFM1 (NM_004208.3; NP_004199.1) was used as the reference sequence for mutation nomenclature.
Clinical manifestations of AIFM1-associated ANSD
The phenotype associated with the AUNX1-causing AIFM1 p.R451Q mutation is characterised by childhood-onset ANSD and delayed peripheral sensory neuropathy presenting as extremity numbness, unsteadiness and areflexia.25 This clinical picture was seen with the other familial cases of AIFM1 ANSD (0223, 7170, 2724 and 2423) and in some patients with sporadic AIFM1 ANSD (table 1 and online supplementary table S9). The electrophysiological findings of 14 nerves in seven affected familial members were obtained (see online supplementary table S11). Nerve conduction studies demonstrated reduced sural, median and ulnar sensory NCVs or even absent responses. The reduced SCVs were always associated with reduced SNAPs (see online supplementary figure S3). The abnormal SEP results were also recorded, including no response or prolonged latency for the evoked potential P40 of tibial SEP, with or without prolonged latency of N9 potential of median SEP (see online supplementary figure S4). The data indicated that the patients might have demyelination changes in the peripheral sensory nerves. However, the MCVs and CMAPs of all patients were normal (see online supplementary figure S3). Needle EMG performed in these patients also showed normal values. There were no fibrillation, positive or fasciculation potentials and myotonic discharges were not observed. The patients also showed normal motor unit action potentials (see online supplementary figure S5). All of these findings suggested that the affected patients had evidence of peripheral sensory neuropathy but not motor neuropathy nor myopathy. The MMSE scores of patients III: 1, III: 9 and III: 11 from family 2423 were in the normal range (27–30): 28, 27 and 28, respectively with their corresponding educational backgrounds of junior college, high school and middle school. These normal scores (≥27) indicated normal cognitive function.
Re-examination of patients showed that both hearing impairment and sensory neuropathy slowly progress.10 ,25 Although serial cerebral MRI in familial ANSD demonstrated normal signal intensity in the brain, inclined sagittal MRI of the internal auditory canals showed bilateral cochlea nerve hypoplasia (CNH), a finding consistent with the diagnosis of ANSD (figure 4A, B and table 1). No symptoms or signs of muscle wasting, weakness or atrophy were identified in AIFM1 mutated patients, and muscle biopsy of the left gastrocnemius in an affected member of family 0223 (III: 2) revealed only a few atrophic myofibers (figure 4C–G). All patients with AIFM1 mutations had normal serum levels of lactate dehydrogenase and creatine kinase.
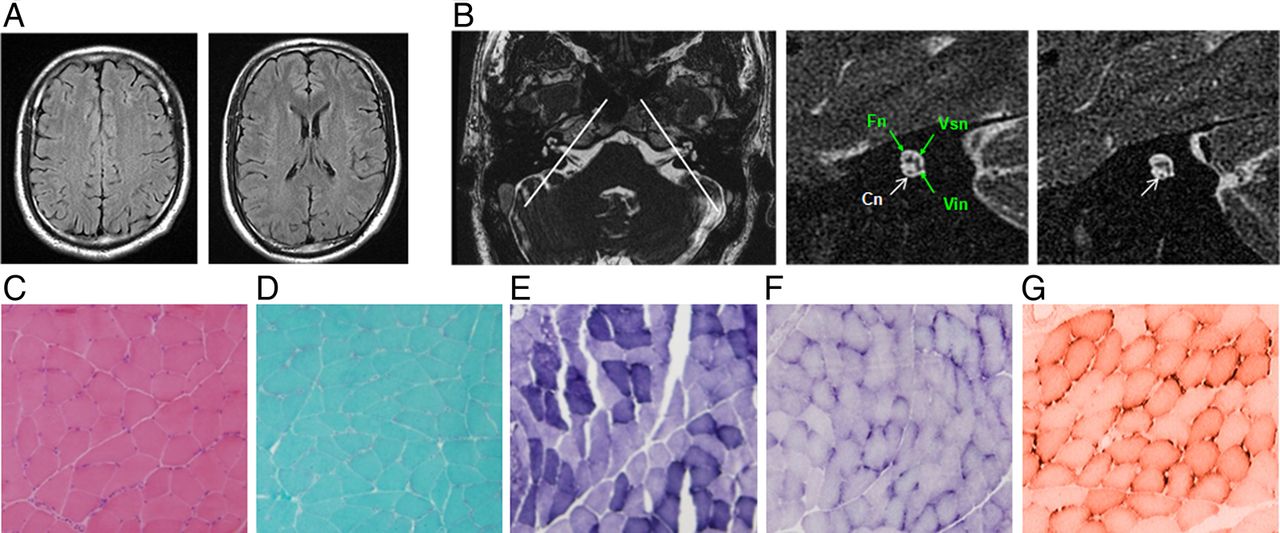
Brain MRI imaging and muscle biopsy immune-staining of the patient (III: 3) from family 0223. (A) Serial cerebral MRI with fluid-attenuated inversion recovery sequence demonstrates normal signal intensity in bilateral centrum semiovale (left panel) and periventricular and subcortical white matter (right panel). (B) Axial view of the cerebellopontine angle and the internal auditory canal (IAC) shows normal anatomy (left panel). The two white lines illustrate the plane prescribed for oblique plane sagittal images obtained perpendicular to the nerves of the IAC. The oblique plane sagittal image (3D-fast-spin echo sequence, middle panel) obtained on the left side demonstrates an abnormally small cochlea nerve (Cn, white arrow) but a normal size IAC with normal facial (Fn), superior (Vsn) and inferior (Vin) vestibular nerves (green arrows). The right Cn was symmetrically small (right panel, white arrow). (C–F) Immunohistochemical staining of muscle biopsy (left gastrocnemius) in patient III: 3 shows a few atrophic myofibers (H&E, C). No ragged red fibres (modified Gomori-trichrome, D), ragged blue fibres (succinate dehydrogenase, E) or targetoid fibres (nicotinamide adenine dinucleotide-tetrazolium reductase, F) are identified. There is no reduction or absence of cytochrome-c-oxidase histochemical reactions observed (G).
Impact on protein structure
We evaluated the functional effects of the 11 amino acid substitutions identified in this study using Polyphen2, SIFT, PROVEAN and Mutation Assessor.26–30 Nine variants were predicted to be likely pathogenic by at least three programs. (We considered the following predictions as pathogenic: Polyphen2, probably damaging; SIFT, damaging; PROVEA, deleterious; Mutation Assessor, medium/high functional impact). The exceptions, p.R422Q and p.I591M, were predicted pathogenic by two and one program, respectively (figure 3 and online supplementary table S12). Structural comparison of wild-type versus mutated AIFM1 protein showed that mutations in the two flavin adenine dinucleotide (FAD) and reduced nicotinamide adenine dinucleotide (NADH) domains have greater impact on the protein surface than mutations in C-terminus (see online supplementary figures S6 and S7).
Histological findings
Immunostaining of murine inner ear demonstrated ubiquitous localisation of AIFM1 in the inner, especially to the cytoplasm of IHC, OHCs and spiral ganglion neurons, consistent with a role in normal auditory function (figure 5 and online supplementary figures S8–S9).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Localisation of AIFM1 in the murine inner ear by immunostaining with a monoclonal AIFM1 antibody. (A) Organ of Corti whole-mount preparation demonstrates AIFM1 (red) localisation to the cytoplasm of inner hair cell (IHC) and outer hair cells (OHCs), as well as the surrounding tissue. (B) Control organ of Corti tissue labelled with only secondary antibody and phalloidin (green). (C) Spiral ganglion whole-mount preparation shows AIFM1 (red) staining in spiral ganglion neurons. (D) Control spiral ganglion tissue labelled with secondary antibody and phalloidin (green). The scale bar indicates 15 µm in panels A and B, and 10 µm in panels C and D. DAPI, 4′,6-diamidino-2-phenylindole dihydrochloride.
Discussion
In 2006, we mapped a novel X-linked auditory neuropathy locus (AUNX1) to chrXq23-q27.3 in a large five-generation Chinese family.10 Using WES and confirmatory segregation analysis, we now report a novel missense change, p.R451Q in AIFM1, as causally responsible for the phenotype in this family. Consistent with its playing a major role in ANSD, we have identified 10 other mutations in AIFM1 in a cohort of familial and sporadic cases of ANSD of Chinese ethnicity. In all familial cases, the identified variants (p.T260A, p.L344F, p.R422W and p.R422Q) co-segregate with the auditory and peripheral sensory neuropathy. All 11 variants were absent in a screen of 500 normal-hearing, ethnicity-matched Chinese controls (750 X chromosomes) and 9 were classified as likely damaging by multiple bioinformatics programs (see online supplementary table S12). In aggregate, these data provide abundant compelling evidence to implicate AIFM1 in X-linked recessive ANSD.
AIFM1 encodes apoptosis-inducing factor 1, a flavoprotein located in the mitochondrial intermembrane space. AIFM1 has at least two functions.31–33 First, as a caspase-independent death effector, it mediates caspase-independent programmed cell death when translocating from mitochondria to the nucleus upon apoptotic stimuli. And second, as an FAD-dependent NADH oxidoreductase, it plays an important role in oxidative phosphorylation, redox control and respiratory chain activity in healthy cells. To date, AIFM1 mutations have been associated with a severe mitochondrial encephalomyopathy (COXPD6, MIM# 300816; caused by p.R201del),11 prenatal ventriculomegaly (caused by p.G308E)12 and Cowchock syndrome (CMTX4, MIM# 310490; caused by p.E493V).13 Common features of these disorders are developmental disabilities such as mental retardation, motor dysfunction and muscle weakness, and abnormal MRI findings in brain.11–13 The AUNX1 phenotype is very different from these other phenotypes, as is the location of the causal mutations in the protein (see online supplementary table S13 and figure S10).
Based on phenotypic variability, it has been suggested that AIFM-related diseases have differing pathogenic mechanisms.34 In Cowchock syndrome, the p.E493V mutation alters the redox properties of the mutated protein, resulting in increased apoptosis.13 In COXPD6, in comparison, the R201del mutation reduces activity of respiratory chain complexes I–V and increases caspase-independent programmed cell death.11 Most of the 11 mutations identified in this study are located in the NADH and second FAD domains of AIFM1, which are essential for FAD-dependent NADH oxidoreductase.
Interestingly, in spite of the widespread expression of AIFM1 in murine inner ear, which is consistent with a role in normal auditory function, the mutated protein did not affect OHCs function as measured by distortion product otoacoustic emission responses (figure 5 and online supplementary table S9). In addition, while some patients with AIFM1 mutations had MRI-documented CNH, the onset of hearing problems was typically during adolescence, suggesting that the hypoplasia represents late-onset and not congenital degeneration (see online supplementary figure S10; table 1).35 ,36
ANSD is known to be an extremely complex disease that has congenital and acquired forms. Extensive clinical testing and genetic research are invaluable to elucidate underlying mechanisms and sites of pathology.37–39 Our finding of bilateral CNH in AIFM1-related ANSD implies that MRI screening may identify the site of lesion in some patients with this phenotype. Furthermore, it suggests that if CNH is an eventual common outcome cochlea implantation in patients with AIFM1-related ANSD may meet with limited success.40–42 In most patients, we were able to diagnose the other aspect of the phenotype, peripheral sensory neuropathy, by clinical and neurophysiological testing, although the symptoms of sensory neuropathy may occur many months or even years after the auditory neuropathy (see online supplementary table S11).
In conclusion, our study identifies AIFM1 as a new causal gene associated with X-linked auditory neuropathy and delayed peripheral neuropathy. These results expand the spectrum of AIFM1-associated diseases to include ANSD. Because female carriers are unaffected, AIFM1 should be considered in small pedigrees with apparent autosomal recessive ANSD if X-linked inheritance cannot be excluded. Further studies are required to determine the long-term benefit these patients may receive from cochlear implantation.
Acknowledgments
We would like to thank the families and the patients for their invaluable cooperation and participation. We thank Chao Zhang and Lan Yu for auditory testing in patients, Cui Zhao and Na Li for collecting and preparing DNA samples, Nan Liu for assistance with PCR and Sanger sequencing.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online figures
- Data supplement 2 - Online tables
Footnotes
LZ, JG, ME contributed equally to this work.
Contributors The first three authors (LZ, JG, ME) contributed equally to this work. Performed the experiments: LZ, JG, ME, QZ, YZ, ZS, FW, JY, CB, WS, LL, DD, LX, YQ, QS and WX. Contributed reagents/materials/analysis tools: DW, HW, WS, DD, XL, XH, DW, SC, ZY, NY, JuW and JiW. Wrote the paper: QW, LZ and RJHS. Critical reading and discussion of manuscript: QW, RJHS, RJS and CP. QW and RJHS are guarantors responsible for this work.
Funding This study was financially supported by the National Key Basic Research Program of China (no. 2014CB943001) and the National Natural Science Foundation of China, Major Project (no. 81120108009) to QW; and the National Institutes of Health (NIDCD DC003544, DC002842 and DC012049 to RJHS). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Competing interests None declared.
Patient consent Obtained.
Ethics approval The Ethics Reviewing Committee of the Chinese PLA General Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.