Article Text

Abstract
Background De novo mutations are emerging as an important cause of neurocognitive impairment, and whole exome sequencing of case-parent trios is a powerful way of detecting them. Here, we report the findings in four such trios.
Methods The Deciphering Developmental Disorders study is using whole exome sequencing in family trios to investigate children with severe, sporadic, undiagnosed developmental delay. Three of our patients were ascertained from the first 1133 children to have been investigated through this large-scale study. Case 4 was a phenotypically isolated case recruited into an undiagnosed rare disorders sequencing study.
Results Protein-altering de novo mutations in PURA were identified in four subjects. They include two different frameshifts, one inframe deletion and one missense mutation. PURA encodes Pur-α, a highly conserved multifunctional protein that has an important role in normal postnatal brain development in animal models. The associated human phenotype of de novo heterozygous mutations in this gene is variable, but moderate to severe neurodevelopmental delay and learning disability are common to all. Neonatal hypotonia, early feeding difficulties and seizures, or ‘seizure-like’ movements, were also common.
Additionally, it is suspected that anterior pituitary dysregulation may be within the spectrum of this disorder. Psychomotor developmental outcomes appear variable between patients, and we propose a possible genotype–phenotype correlation, with disruption of Pur repeat III resulting in a more severe phenotype.
Conclusions These findings provide definitive evidence for the role of PURA in causing a variable syndrome of neurodevelopmental delay, learning disability, neonatal hypotonia, feeding difficulties, abnormal movements and epilepsy in humans, and help clarify the role of PURA in the previously described 5q31.3 microdeletion phenotype.
- Clinical genetics
- Developmental
- Epilepsy and seizures
- Neurology
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Neurodevelopmental disorders are common and encompass a broad range of intellectual, behavioural and motor disabilities. Learning disability alone affects 1%–3% of the population and, for the most part, has a complex genetic basis.1 Indeed, it is this complex genetic heterogeneity and variability of expression that has previously posed a significant barrier to the investigation and molecular diagnosis of neurodevelopmental disorders.
However, with the advent of next-generation sequencing technology, extensive interrogation of the exome has become possible. The use of whole exome sequencing (WES) has enabled the identification of pathogenic mutations in patients with well-characterised neurodevelopmental phenotypes, such as Kabuki syndrome2 and Schinzel–Giedion syndrome.3
In many cases, however, there may be no consistent physical characteristics to help group patients with sporadic neurodevelopmental disorders for molecular genetic investigation. For this reason, such cases are inherently more challenging. One paradigm that has proved to be extremely effective is WES in family trios.4 ,5 This approach has helped to successfully identify numerous pathogenic de novo mutations as the cause of sporadic neurodevelopmental delay,4 ,6 ,7 and forms the basis of the Deciphering Developmental Disorders (DDD) study, through which the mutations in three of our four patients were identified.
The enrichment for de novo mutations as a cause of sporadic neurodevelopmental disorders is not surprising given the overall association with reduced fecundity and the baseline rate of DNA replication errors, which has been reported from detailed genomic studies as ∼10−8 de novo germline base substitutions per base pair per generation.8
We report four unrelated children with significant neurodevelopmental delay who have been investigated by WES in family trios and found to have pathogenic de novo mutations in PURA (MIM 600473).
Methods
Of our four patients, three were referred to regional Clinical Genetics services across the UK, where they were recruited to the DDD study (http://www.ddduk.org). DDD has so far investigated 1133 children with severe, undiagnosed developmental delay, and their parents, using a combination of genome-wide assays to detect all major classes of genetic variation in the protein-coding portion of the genome. They have recorded clinical information and phenotypes using the Human Phenotype Ontology9 via a secure web portal within the DECIPHER database.10
DNA samples from patients and their parents were analysed by the Wellcome Trust Sanger Institute using high-resolution microarray analysis (array-comparative genomic hybridisation (CGH) and SNP-genotyping) to investigate CNVs in the child, and exome sequencing to investigate SNPs and small insertions/deletions (indels). Putative de novo sequence variants were validated using targeted Sanger sequencing. The population prevalence (minor allele frequency) of each variant in nearly 15 000 samples from diverse populations was recorded, and the effect of each genomic variant was predicted using the Ensembl Variant Effect Predictor.11 Likely diagnostic variants in known developmental disorder genes were fed back to the referring clinical geneticists for validation and discussion with the family via the patient's record in DECIPHER, where they can be viewed in an interactive genome browser. Full genomic datasets were also deposited in the European Genome–Phenome Archive (http://www.ebi.ac.uk/ega).
Patient 4 was referred to paediatric neurology. She underwent extensive neurological and metabolic investigations in Australia. The exomes of Patient 4 and both parents were sequenced in an n=1 family trio study used for diagnostic exploration by Ambry Genetics using SureSelect Target Enrichment System (Agilent Technologies) followed by 2×100 nt paired-end sequencing on a Illumina HiSeq 2000. Raw sequence reads for Patient 4 and her parents were aligned to the reference human genome (GRCh37), and pedigree-informed variant calling was performed using the Real Time Genomics integrated analysis tool rtgFamily V.3.2.12 All variants were annotated using SnpEff V.3.413 using data from dbNSFP2.414 and dbSNP138.15 Subsequent analysis and identification of candidate variants was performed with an in-house workflow incorporating the annotated variant data and pedigree information.
Results
Clinical photographs of the patients are shown in figure 1. Clinical features are shown in detail in table 1.
Clinical phenotype descriptions of the four index patients
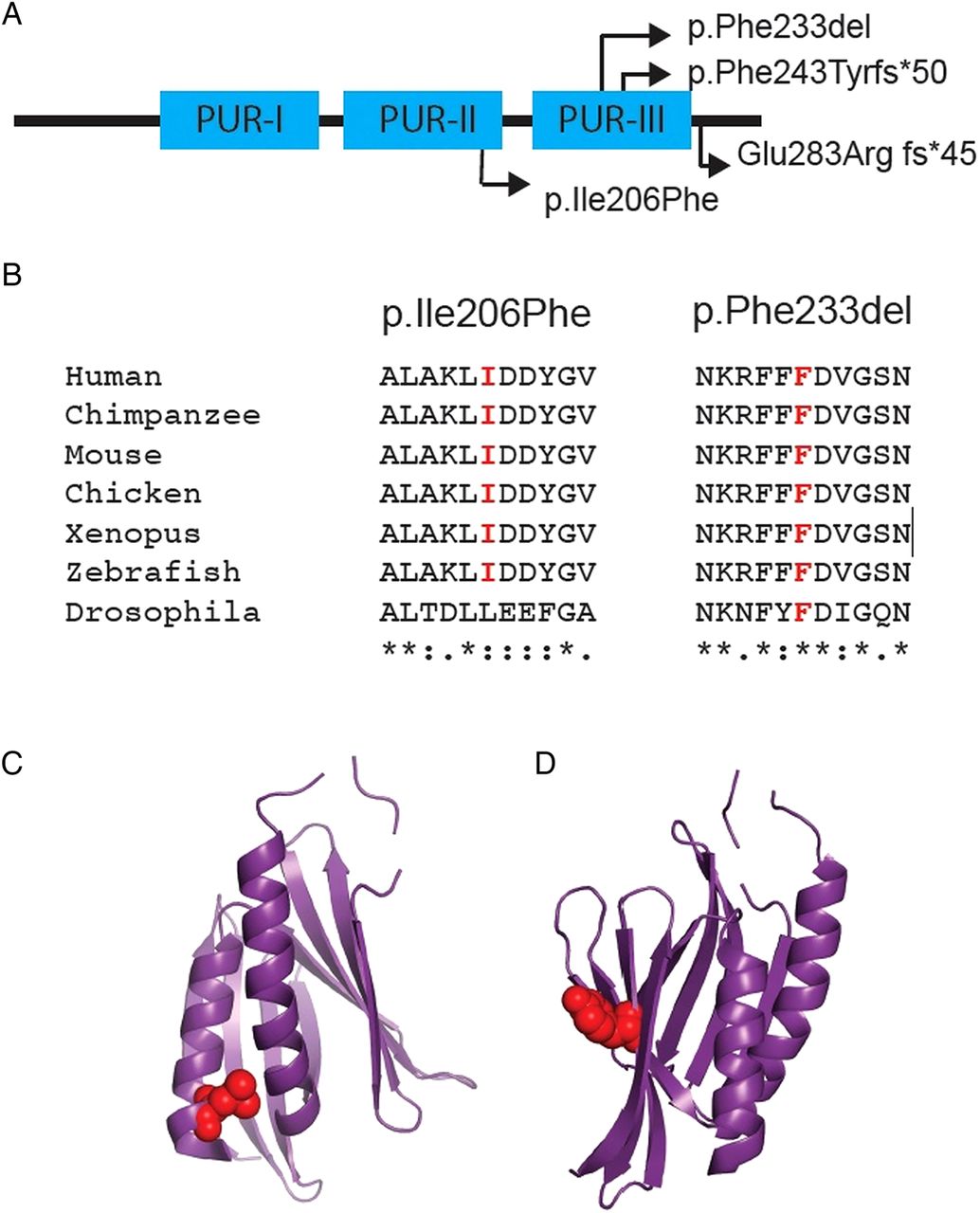
Depictions of the four mutations identified within PURA. (A) Schematic of Purα, depicting sites of the different mutations identified in our patients with respect to the Pur repeat regions (I–III). (B) Sequence alignment illustrating the high level of conservation of amino acids affected by the p.Ile206Phe and p.Phe233del mutations. Asterisk denotes complete conservation, colon denotes high conservation and single dot denotes moderate conservation. (C) and (D) illustrate the respective locations of the p.Ile206Phe and p.Phe233del mutations within the tertiary structure of the protein.
The mutations found by WES for each case are described below. No other causative mutations were indentified in the exomes.
Patient 1
Patient 1 has a de novo frameshift mutation (p.Phe243Tyrfs*50) in PURA.
She had neonatal hypotonia and was nasogastric (NG) tube-fed for the first week of life. Her swallow has remained poor. A single apnoeic/hyponoeic episode occurred while she was a neonate. Abnormal ‘seizure-like’ movements were investigated at 7 months of age by EEG, which was normal. Visual evoked potentials revealed broadened wave forms consistent with neurological nystagmus (with preserved optokinetic nystagmus).
At her last clinical assessment, aged 4 years 7 months, she was not walking, and remained non-verbal. Patient 1 had hypotonic facies with a prominent forehead, epicanthic folds and mild telecanthus. Lower limb posture was abnormal with feet held in plantar flexion. There was restricted ankle movement, mild hypotonia and generalised weakness. Coordination was poor, but not grossly ataxic. Intermittent dysconjugate gaze was noted.
Prominent early breast bud development led to endocrine investigations that revealed this to be gonadotropin-dependent. She has been treated with intramuscular decapeptyl from age 3 years. MRI brain scans show delayed myelination.
Patient 2
Patient 2 has a de novo frameshift mutation (p.Glu283Argfs*45) in PURA.
She did not have neonatal hypotonia, respiratory difficulties or feeding problems. She achieved unsupported sitting at 12 months, independent walking at 24 months and first words at 2 years 6 months. At 14 years 3 months, she was independently mobile, able to dress herself and feed herself. She communicates in sentences, although with limited vocabulary. Patient 2 has an anxious disposition and lacks awareness of danger. Clinical examination revealed microcephaly, tall forehead, hypotonic facies, mild facial asymmetry, upslanting palpebral fissures and large central incisors. She has long thin fingers and toes, with 5th finger clino/camptodactyly bilaterally, over-riding 2nd toes and deep palmar creases.
Patient 3
Patient 3 has a non-synonymous missense mutation (p.Ile206Phe). This amino acid substitution arises in Pur repeat II, a very highly conserved region of sequence within Purα (figure 2). Crystallography studies suggest that Pur repeat I and Pur repeat II interact to form a functional Pur domain.16 In silico analysis with SIFT and PolyPhen produced scores of 0.01 and 0.969, respectively, supporting pathogenicity.

Clinical photographs. Patient 1 is shown at age 1 year 7 months (A and B) and 4 years 7 months (C). Patient 2 is shown at 7 years 3 months (D and E) and 14 years 3 months (F). Patient 3 is shown at 9 years 7 months (G and H) and 12 years 10 months (I). Patient 4 is shown at 6 years 9 months (J and K). While there is no obvious gestalt, all four patients were noted to have quite prominent foreheads with high anterior hairlines. Patients 1, 2 and 4 have mildly hypotonic facies.
Respiratory distress at birth necessitated supplementary oxygen. Neonatal hypotonia and hypoglycaemia were present, and NG tube feeding was necessary. She sat unsupported at 12–14 months, walked at 22 months and said her first words at ∼3 years 6 months. At 12 years 10 months, she was able to run unsteadily with a wide-based gait. She communicates at a basic level with short phrases, repetitive speech and limited comprehension. Her behaviour can be obsessional and attention-seeking, with limited awareness of others. Patient 3 has a long face, full cheeks, high forehead and telecanthus. Neurological examination revealed hypotonia, mild weakness and poor coordination. EEG at 3 years for possible seizures showed: occasional paroxysmal discharges in the form of spikes and sharp waves over the right frontal and left mid-temporal region in sleep.
Patient 4
Patient 4 has an inframe deletion (p.Phe233del), affecting a very highly conserved phenylalanine residue within Pur repeat III, a presumed functional domain of Purα which is necessary for homodimerisation in crystallography studies16 (figure 1), and is present even in very distantly related organisms such as Caenorhabditis elegans. It is, therefore, highly likely to be of functional significance.
She developed central apnoea, hypothermia and severe hypotonia from day 2 of life. There was absent suck and gag reflex requiring early NG feeding. At 6 years 9 months, unsupported sitting had not been achieved. There is little language development.
She has hypotonic facies, frontal bossing and thin upper lip. Neurological examination revealed generalised hypotonia and dystonic/dyskinetic facial and limb movements. There was generalised weakness but no ataxia. Eye movements were dysconjugate.
Seizures commenced at 14 months with infantile spasms, and progressed to tonic seizures and focal dyscognitive seizures. Seizures have proved difficult to control.
EEG recordings have been normal or mildly slow when seizures are under control, but highly abnormal during seizures with near-continuous multifocal and bisynchronous sharp/slow activity maximal posteriorly. Video telemetry has revealed epileptic spasms and/or tonic seizure activity. Inborn errors of neurotransmitter biosynthesis and metabolism have been excluded by both Sanger and WES.
Serial MRI brain scans have been performed since birth (figure 3). These showed a right frontal horn cyst, which subsequently resolved. There was also patchy high attenuation throughout the white matter. Myelination was delayed but complete by 5 years, by which time there was evidence of excessive extra-axial fluid spaces and possible parenchymal volume loss. MR spectroscopy has demonstrated decreased N-acetyl aspartate within the frontal lobes and basal ganglia.

{kind=link}
{kind=link}
{kind=link}
Serial MRI brain scans from Patient 4. (A) At 1 week, there is patchy high attenuation within white matter and a right frontal horn cyst, which is not evident on subsequent scans. (B) At 14 months, the white matter appears normal but thickening of tissue at the ependymal margin of the right frontal horn is apparent. (C) At 2 years 2 months, subtle hypomyelination is apparent in that there is poor definition of the grey-white matter boundary in the frontal lobes. (D) At 3 years 10 months, subtle hypomyelination persists. (E) At 5 years, myelination is complete. However, there are excessive extra-axial fluid spaces and there is possible cerebral atrophy.
Discussion
PURA encodes a ubiquitously expressed protein, Purα, which contains an N-terminal glycine-rich region, three Pur repeats (I–III) and a C-terminal glutamine–glutamate rich domain16 (figure 1). The full-length protein is 322aa in humans and gives rise to a 28 kDa product.17 ,18
Purα is very highly conserved across the phylogenetic tree (figure 1), with regulatory roles in DNA replication, gene transcription, RNA transport and mRNA translation. Originally, it was identified in mouse due to its ability to bind to a sequence within the myelin basic protein promoter.19 ,20 The human form was identified through its binding to a purine-rich element within an origin of DNA replication upstream of the human c-MYC gene.17 A consensus sequence for the purine-rich single strand of the so-called PUR element, to which Purα binds, was subsequently derived. It has since become apparent that Purα's preferential recognition sequence comprises GGN repeats.
In order to initiate DNA replication and gene transcription, Purα first destabilises the DNA helix so that it may then bind its target sequence on a single DNA strand.21 It is able to bind both linearised and supercoiled DNA. However, mutation studies have revealed that the carboxy terminal segment of Purα, which includes Pur repeat III, is necessary for destabilisation of linearised DNA.22
Purα has been shown to be important in controlling gene transcription from an array of different genes. Interestingly, it has gene-specific roles as either an activator or repressor of transcription. Purα activates transcription for a large number of cellular genes including those encoding myelin basic protein,19 tumour necrosis factor α,23 BC124 and the neuron-specific TATA-less gene FE65.25 Furthermore, proteomics studies suggest that both Purα and its paralog, Purβ, may have an important regulatory role in control of the gene expression of myelin proteolipid protein (Plp1), which is the most abundant protein in central nervous system myelin and is developmentally regulated. Expression of Plp1 peaks in oligodendrocytes during active myelination.26 By contrast, Purα represses expression from a wide range of genes including amyloid-β precursor protein,27 α-actin28 and gata2.29 There is also evidence that Purα is involved in controlling its own transcription through a process of autoregulation.30
There are two independently generated Pura−/− mice which have helped our understanding of Purα's role in normal development.31 ,32 Both knockout mice are reported to appear normal at birth and develop neurological features at approximately 2 weeks of age, which include continuous and increasingly severe tremor. Khalili et al31 reported that their mice appeared to feed well but did not gain weight normally and died at 1 month of age. They also noted that their heterozygous mice were prone to seizures on routine handling. Hokkanen et al32 reported that their null mutant mice lived up to 6 months. They reported that these animals did not gain weight normally after onset of tremor. They also observed an ataxic gait in these animals with an apparent hind limb weakness.
Both groups found that there was a marked reduction in the expression of the dentritic protein MAP2. This is interesting because Purα binds to mouse BC1 RNA in complex with other proteins such as Fragile X Mental Retardation Protein and Staufen, as well as various mRNA species.33 These form so-called messenger ribonucleoprotein granules, which are critical to normal dendritic function.
Purα binds to the (CGG)n sequence in FMR1 that is pathologically expanded in Fragile X syndrome. Intriguingly, it has been suggested that Fragile X-associated tremor/ataxia syndrome (FXTAS), which may arise in premutation carriers, is due to the sequestration of Purα and other rCGG repeat binding proteins, thereby preventing them from fulfilling their normal cellular function.34 The movement disorder observed in knockout mice might therefore be functionally related to FXTAS.
Until now, there have been no specific reports of mutations within PURA as a cause of human disease. It is, however, noteworthy that a 5q31.3 microdeletion phenotype has recently emerged35 and PURA, which lies within the shared deletion interval of the seven patients described to date, and has been proposed as a candidate gene for the associated phenotype.36 ,37 The shared phenotype of all patients reported thus far includes hypotonia, feeding difficulty and developmental delay. Additionally, respiratory problems, such as apnoea, and seizures or ‘seizure-like’ movements are reported in the majority of these patients.
While NRG2, a member of the neuregulin family, is highly likely to be contributory to the 5q31.3 microdeletion phenotype,36 one of the two most recent patients to have been described in the literature with a similar phenotype has a microdeletion that has narrowed down the shortest region of overlap (SRO) to a 101 kb region encompassing only three genes: PURA, C5orf53 and C5orf32. Given that the function of the latter two genes is yet to be characterised, Brown et al37 have proposed that this ‘lends further support for PURA as the likely primary candidate gene for the core neurodevelopmental features of this (5q31.3 microdeletion) syndrome’.
In this report, we provide the first evidence, that mutations limited to PURA are indeed sufficient to cause significant neurodevelopmental delay and learning disability in humans.
The four unrelated index patients have different de novo mutations in PURA. Patients 1 and 2 have frameshift mutations (p.Phe243Tyrfs*50 and p.Glu283Argfs*45, respectively). Given that PURA is a single exon gene, these altered gene products would not be subject to nonsense-mediated decay. As such, there is potential for these translated proteins to have dominant negative or gain-of-function effects or, alternatively, result in functional haploinsufficiency. Patient 3 has a missense mutation (p.Ile206Phe) and Patient 4 has an inframe deletion (p.Phe233del). Both of these mutations occur within highly conserved regions of sequence, giving rise to the Pur repeat II and Pur repeat III regions, respectively. These repeat regions are unique to Purine-rich element-binding proteins, and are of functional significance.
In all four affected individuals (figure 1), there was a shared core phenotype (table 1) of moderate to severe neurodevelopmental delay. Central hypotonia and early feeding difficulties were also common, as were respiratory difficulties ranging from distress at birth to single or recurrent central hyponoeic/apnoeic episodes in the newborn period. Three of our patients have a history of seizures or ‘seizure-like’ movements.
Additionally, some unusual features have been noted that may be part of the phenotypical spectrum for PURA mutations. In particular, there are some notable endocrine problems among these patients. Patient 1 has a history of gonadotropin-dependent precocious puberty with persistently elevated luteinizing hormone and follicle-stimulating hormone. She has early breast bud development and is currently on treatment with decapeptyl. None of the other patients are reported to have signs of early puberty. However, Patient 4 does have a history of other endocrine abnormalities including a persistently raised prolactin in the neonatal period and a blunted cortisol response to stress, despite normal baseline levels. She also has persistently low vitamin D levels despite treatment. This suggests that there may be a wider endocrine component, particularly with respect to anterior pituitary function.
Intriguingly, there is some evidence that Purα may be involved in the regulation of gondatropins. One study seeking to identify novel DNA-binding proteins for gonadotropin-releasing hormone 1 (GNRH1) promoter, identified both Purα and Purβ as potential regulators of GNRH1 gene expression.38 Subsequent in vivo studies have confirmed binding of both Purα and Purβ to the upstream region of the GNRH1 gene. While overexpression of Purβ was shown to significantly downregulate GNRH1 expression in transiently transfected mouse GT1-7 cells, this could not be demonstrated for Purα. However, it is worth noting that there is evidence that Purα is able to form a functional heterodimer with Purβ.39
Curiously, only Patient 1, the youngest, had a head circumference that appeared to be growing at the expected rate. None of the other 3 patients have maintained their projected rate of head growth from early occipital-frontal circumference measurements. This presumably reflects an inadequate growth in underlying brain volume, although no discrete brain structures were noted to be hypoplastic on MRI. This apparent inability to maintain growth velocity is consistent with the observations made in Pura−/− mice by Khalili et al.31
MRI brain scans were performed on all four patients at various ages. Patients 2 and 3 had normal MRI brain scans at ages 7 and 10 years, respectively. Historical scans were not available to check for early evidence of delayed myelination in these patients. In Patient 1, delayed myelination was detected at 3 years 5 months. In Patient 4, serial MRI brain scans were performed from birth showing a number of abnormalities including a transient right frontal horn cyst and patchy high attenuation of the white matter at birth and delayed myelination, with myelination complete by 5 years of age. There were, however, enlarged extra-axial fluid spaces by this time raising the possibility of mild parenchymal volume loss. As such, it is reasonable to say that evidence of delayed myelination was found only in those patients whose scans were performed early enough to detect it. On the whole, our patients’ brain imaging is not entirely typical of the findings reported in the 5q31.3 phenotype, which includes frontotemporal volume loss, simplified frontal gyral pattern with shallow sulcation and delayed or incomplete myelination of the frontotemporal subcortical white matter tracts and anterior limbs of the internal capsules and cyst formation. However, Patient 4's brain imaging bears the greatest overlap with this phenotype.
We are confident that all four patients’ phenotypes are secondary to their de novo mutations in PURA. WES has excluded other significant gene mutations. Furthermore, array CGH has excluded chromosomal microdeletions or duplications that may not necessarily have been detected by WES alone. Additionally, all patients have been thoroughly investigated by multiple physicians of various specialities en route to their definitive molecular genetic diagnosis. While there is a core phenotype, there is variability among our patients. Further cases will be useful to assess any distinctive genotype–phenotype correlations, or whether features, such as endocrine disturbance, metabolic abnormalities, epilepsy and a movement disorder might represent rare manifestations within a broad phenotypical spectrum. Regardless, our assumption has been that a functional haploinsufficiency has resulted from all four mutations. Functional studies may be necessary to confirm this hypothesis and exclude other possibilities, such as dominant negative or gain-of-function effects. However, the modular architecture of Purα and functional studies that have been completed to date tell us that truncating frameshift mutations similar to those found in Patient 1 will almost certainly have abolished the functional Pur repeat III sequence that is necessary for dimerisation and binding to linearised DNA. Patient 4's inframe deletion affects a very highly conserved residue within the same Pur repeat and would be expected to cause similar functional problems. Both these children are severely affected, being non-ambulatory and non-verbal. They are, however, the youngest two patients—but they have already exceeded the ages at which Patients 2 and 3 achieved independent ambulation (22–24 months).
Patient 2's frameshift mutation is downstream of the Pur repeats. The functional effect is not clear at a molecular level, but it seems to be associated with a less severe neurodevelopmental phenotype. Patient 3's missense mutation falls with Pur repeat II and affects a highly conserved residue. Again, the functional effect at a molecular level is not yet clear, but it presumably has potential to interfere with the formation of the ssDNA/ssRNA binding domain. Regardless, it too appears to be associated with a less severe neurodevelopmental phenotype.
We believe that our four patients help to resolve the 5q31.3 microdeletion phenotype. In the Brown et al37 study, Patient 2, whose deletion significantly narrowed the SRO, was more mildly affected than the other six patients whose 5q31.3 microdeletions also included NRG2 (MIM 603818). Additionally, this patient is reported as non-dysmorphic, whereas the other six patients have quite strikingly dysmorphic features. It has, therefore, been suggested that the combined deletion of PURA and NRG2 (and/or other genes within the SRO for these six patients), may account for a more severe phenotype.37 It has also been suggested that the more dysmorphic appearance of these patients is, in part, due to their more profound state of hypotonia. Our findings support the hypothesis that the deletion of PURA contributes to, but is not the sole cause of, the 5q31.3 microdeletion phenotype.
With the exception of mildly hypotonic facies, which are apparent in three of our patients, there are no obvious consistent dysmorphic facial features in this first cohort. We note, however, that all four patients have fairly prominent foreheads with relatively high anterior hairlines. As such, this is not a genetic syndrome that currently lends itself readily to clinical diagnosis based on history and clinical examination findings alone. However, if in time there should prove to be clear associations with discrete clinical problems, such as gondatropin-dependent precocious puberty or consistent brain imaging findings, it may be that the diagnosis can be strongly suspected on clinical grounds. However, based on the patients described herein, we suspect that this will ultimately prove to be a diagnosis that is usually made following investigation by WES or gene panel testing for neurodevelopmental delay. Indeed, as such technology becomes more readily accessible to clinicians, this diagnosis will undoubtedly become recognisable as a rare but important cause of sporadic neurodevelopmental delay.
Acknowledgments
The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (grant number HICF-1009-003), a parallel funding partnership between the Wellcome Trust and the Department of Health, and the Wellcome Trust Sanger Institute (grant number WT098051). The views expressed in this publication are those of the author(s) and not necessarily those of the Wellcome Trust or the Department of Health. The research team acknowledges the support of the National Institute for Health Research, through the Comprehensive Clinical Research Network. We would like to thank Associate Professor Avihu Boneh and Dr Diana Johnston who contributed clinical data for Patient 4. We thank the patients and their families for their participation. DB is a Hefce senior clinical fellow.
References
Footnotes
-
Contributors All the authors contributed significantly to this research and preparation of the manuscript. DH is the first author and along with DB (corresponding and senior author) coordinates the group. DB, ACM, PDT and DH phenotyped the UK clinical cases, put forward the sample trios for exome through the DDD study and interpreted the results obtained. RJL and KS phenotyped the Australian case, with CS and RT undertaking the exoming and interpretation of results. MG-C assessed the clinical findings of all the cases radiologically. All authors have been involved in the drafting, critical revision and final approval of the manuscript for publication. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC), Royal Children's Hospital Melbourne Human Research Ethics Committee approval (REF: 28097 G) and The University of Queensland Human Research Ethics Committee approval (2013001536).
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Web Resources Online Mendelian Inheritance in Man, http://www.omim.org/; Simple Modular Architecture Research Tool (SMART), http://smart.embl-heidelberg.de; UCSC Genome Browser, http://genome.ucsc.edu
-
Sequence Data Coding sequence mutations in PURA are referenced against NM_005859.4.