Article Text

Abstract
Background Subtelomeric deletions and duplications may cause syndromic disorders that include features of immunodeficiency. To date, no phenotype of immunological pathology has been linked to partial trisomy 19. We report here on two unrelated male patients showing clinical and laboratory signs of immunodeficiency exhibiting a duplication involving Chromosome 19p13.
Methods Both patients underwent a detailed clinical examination. Extended laboratory investigations for immune function, FISH and array comparative genome hybridization (CGH) analyses were performed.
Results The reported patients were born prematurely with intrauterine growth retardation and share clinical features including neurological impairment, facial dysmorphy and urogenital malformations. Array CGH analyses of both patients showed a largely overlapping terminal duplication affecting Chromosome 19p13. In both affected individuals, the clinical course was marked by recurrent severe infections. Signs of humoral immunodeficiency were detected, including selective antibody deficiency against polysaccharide antigens in patient 1 and reduced IgG1, IgG3 subclass levels and IgM deficiency in patient 2. Class-switched B memory cells were almost absent in both patients. Normal numbers of T cells, B cells and natural killer cells were observed in both boys. Lymphocytic proliferation showed no consistent functional pathology, however, function of granulocytes and monocytes as assessed by oxidative burst test was moderately reduced. Moreover, natural killer cytotoxicity was reduced in both patients. Immunoglobulin substitution resulted in a decreased number and severity of infections and improved thriving in both patients.
Conclusions Partial trisomy 19p13 represents a syndromic disorder associating organ malformation and hitherto unrecognised immunodeficiency.
- syndrome with primary immunodeficiency (PID)
- hypogammaglobulinemia
- subtelomeric microduplication and microdeletion
- 19p
- array CGH
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
- syndrome with primary immunodeficiency (PID)
- hypogammaglobulinemia
- subtelomeric microduplication and microdeletion
- 19p
- array CGH
Introduction
Numerical chromosomal aberrations have long been known to be potentially associated with primary immunodeficiency. Well-known syndromes include trisomy 21 as well as Turner's syndrome (45, X0) (reviewed in1). In addition, a number of chromosomal microdeletion syndromes have been described with known immunodeficiency phenotype, including DiGeorge phenotypes associated with 22q11.2 or 10p13-14 microdeletion1 ,2 or Wolf-Hirschhorn syndrome (4p partial deletion).1 ,3
By contrast, microduplication events have rarely been associated with immunodeficiency. Microduplications within the short arm of chromosome 19 are rare: between 1980 and 2010, a total of 10 patients with partial trisomy 19p13 have been reported.4–13 We here present an extension of the phenotype of partial trisomy 19 involving Chromosome 19p13. Two unrelated patients of non-consanguineous background with known syndromic features associated with partial trisomy 19 additionally displayed a marked immunodeficiency status prompting immunoglobulin (Ig) substitution therapy. In syndromic disorders, obvious organ malformation or dysfunction may be the leading clinical phenotype and thus mask immunodeficiency.
Methods
For Methods including cytogenetics please see the online supplementary material.
Patient presentation, clinical course and immunological investigations
We report on two independent patients from two different University Clinics in Austria, whose similarities were noticed in the context of an ongoing collaboration within the Austrian Society of Pediatric Immunology.
Birth, anatomical malformations and growth
Patient 1, a 7-year-old boy was born prematurely in the 32nd week of gestation after an uneventful pregnancy (32+2 weeks, intrauterine growth retardation (IUGR), caesarean section, oligohydramnion) with 990 g body weight (<3%), 34 cm body length (<3%), and a head circumference of 26.5 cm (<3%); his APGAR score was 8/9/9; he was discharged from neonatal intensive/intermediate care at the chronological age of 8 weeks. He is the second child to non-consanguineous healthy parents of Turkish descent aged 34 years (mother) and 40 years (father). The second patient, a now 2-year-old boy, was born prematurely as the second child of a 29-year-old Austrian mother and a 35-year-old Austrian father after an uneventful (third) pregnancy at 31+1 weeks of gestational age (IUGR, caesarean section, APGAR 7/8/9, 660 g birth weight (<3%), 34 cm length (<3%), 26.5 cm head circumference (<3%)); dismissed from neonatal intensive/intermediate care at 3 months of chronological age (corrected gestational age: 42+6 weeks). Growth parameters, weight and head circumference of both patients have remained below the third centile, recent measurements are presented in table 1. The parents and siblings of both patients are healthy, and the extended family histories are negative for syndromic disorders or immunodeficiency.
Genetic and clinical synopsis of two patients with partial trisomy 19 (19p13.3)
Upon birth, both patients showed visible organ malformations including microcephalus, facial dysmorphism including flat nasal bridge, telecanthus, short upslanting palpebral fissures, low set ears, long philtrum, narrow lips, micrognathia (figure 1A,C), as well as urogenital malformations, a perineal hypospadia in patient 1 (figure 1B) and glandular hypospadia, horseshoe kidney, cryptorchism (table 1), camptodactylia on both feet of toes II, III, IV and overlapping of right third/fourth toes (figure 1D) in patient 2.

Images of phenotypical details. (A) facial dysmorphy and (B) perineal hypospadia in patient 1. (C) facial dsymorphy including long philtrum and narrow lips and (D) syndactylia of right third/forth toes in patient 2.
Clinical course and neurodevelopmental status
In his 1st year of life, patient 1 suffered from a bilateral incarcerated inguinal hernia and thus had to undergo surgery. Additionally, he showed developmental hip dysplasia type IVa, which was surgically treated by Pemberton's acetabuloplasty. At the age of 5 years, hypospadia required surgery whereupon a penoscrotal fistula was detected that could not be fully corrected.
An extensive neurological examination in the 2nd year of life (chronological age: 18 months) showed profound neurological impairment and motor plus mental retardation. Neuropaediatric examination revealed muscular hypotonia, reduced muscle strength, brisk deep tendon reflexes, negative Babinski sign; furthermore, at 18 months the boy was unable to sit, to crawl, nor to stand free; and, without further specification, the neurological report mentions showed reduced fine motor skills and speech/language retardation., Testing of the sensory system and cerebellar function was unfeasible. Moreover, a strabismus convergens alternans of the left eye was noted at the age of 18 months. Additionally, moderate bilateral sensorineural hearing loss was detected by brainstem evoked response audiometry (summarised in table 1). Cranial MRI showed a hypoplastic inferior vermis and a slightly extended temporal lobe (data not shown). Furthermore, the patient suffered from EEG-verified epilepsy (habitual primarily generalised seizures, EEG pathology III°) treated with valproate, which resulted in decrease of frequency of seizures (not shown). Later neurological evaluations focused on the antiepileptic treatment, while ongoing physiotherapy and ergotherapy have been aiming to improve the psychomotor situation. Dysphagia prompted the implantation of a percutaneous endoscopic gastrostomy tube at the age of 6 years. Because of a severe osteopenia (z score: −4.6 (0.26/cm3)) resulting in a pathological fracture at the age of 6 years, he is treated with bisphosphonate intravenously (18 mg) every 3 months.
Similar to patient 1, patient 2 suffered from inguinal hernia leading to bilateral herniotomy at 3½ months of age. He was noted to have a persisting ductus arteriosus with marginal haemodynamic relevance. Because of delayed motor and mental development (see below), progressive failure to thrive and feeding problems, intensive physiotherapy and ergotherapy was provided, additional nasogastric tube feeding with formula diet was initiated at 8 months and percutaneous gastrostomy was performed at 20 months. Like patient 1, he is neurologically and intellectually impaired with profound psychomotor retardation and muscular hypotonia (eg, sitting and belly sliding/crawling only at 2 years of age; grabbing a spoon without ability to use it), speech delay (vocalising only syllables like ‘da’ at 2 years), pharyngeal instability, microcephalus, had neonatal seizures treated with barbiturate for 10 days and two generalised seizures in the context of suspected meningitis leading to temporal antiepileptic treatment with levetiracetam, from 18 months to 21 months of age. Denver test analysis at 2 years of age revealed social contact abilities corresponding to 15 months and motor skills corresponding to 6–9 months. Basal EEG activity was found to be mildly abnormal without focal activity, and in CNS-MRI profound atrophy with multiple diffuse white matter lesions and microcystic alterations of the basal ganglia were detected (table 1). Additionally, patient 2 suffers from hypermetropia and divergent concomitant strabismus, largely resembling patient 1; however hearing tests were normal.
Clinical signs of immunodeficiency and immune dysregulation
Both patients fulfilled the classical and revised criteria of a primary immunodeficiency disorder with frequent infections14–17 and showed laboratory signs of immune dysregulation. Starting from the age of 3 weeks after birth, patient 1 developed recurrent invasive bacterial infections including episodes of sepsis; a proven Staphylococcus capitis sepsis at the age of 1½ years was followed by a severe urosepsis at 3 years. Apart from two episodes of pneumonia, chronic relapsing productive bronchitis with laboratory signs of bacterial infections recurred more than eight times per year, requiring inpatient admittance and intravenous antibiotic treatment. Moreover, the boy suffered from recurrent urinary tract infections possibly related to the hypospadia, with microbiological evidence of extended-spectrum beta lactamase (ESBL)-positive-positive Escherichia coli infection on two instances. Considering the clinical history of repeated severe bacterial infections in this patient, laboratory investigations revealing selective antibody deficiency and decreased numbers of class-switched B cells (see below),18 he was started on intravenous immunoglobulin (0.4 g/kg, at monthly intervals) at 5 years of age. This resulted in a significant decrease in the occurrence of infections, especially of the upper respiratory tract and lower respiratory tract.
Patient 2 suffered from relapsing viral bronchiolitis and required frequent hospitalisations for intravenous antibiotics because of recurrent severe infections (invasive pneumonia, frequent episodes of febrile otitis media, bronchitis with bacterial superinfection, two episodes of suspected meningitis with seizures required intensive care (see table 1). Upon immunological investigations hypogammaglobulinaemia was diagnosed and immunoglobulin substitution commenced according to evidence-based guidelines for the treatment of primary antibody deficiencies.18 Intravenous immunoglobulin treatment was used from 20 months of age (0.4 g/kg monthly) and switched to subcutaneous Ig substitution (SCIG; 160–200 mg/kg weekly), resulting in reduced frequency and severity of infections and improved thriving. Recently, patient 2 developed protracted enteropathy with calprotectin concentrations in stool >1800 µg/g (normal <50 µg/g), which may potentially indicate inflammatory bowel disease and awaits further evaluation by endoscopy and histology.
Immunological laboratory investigations
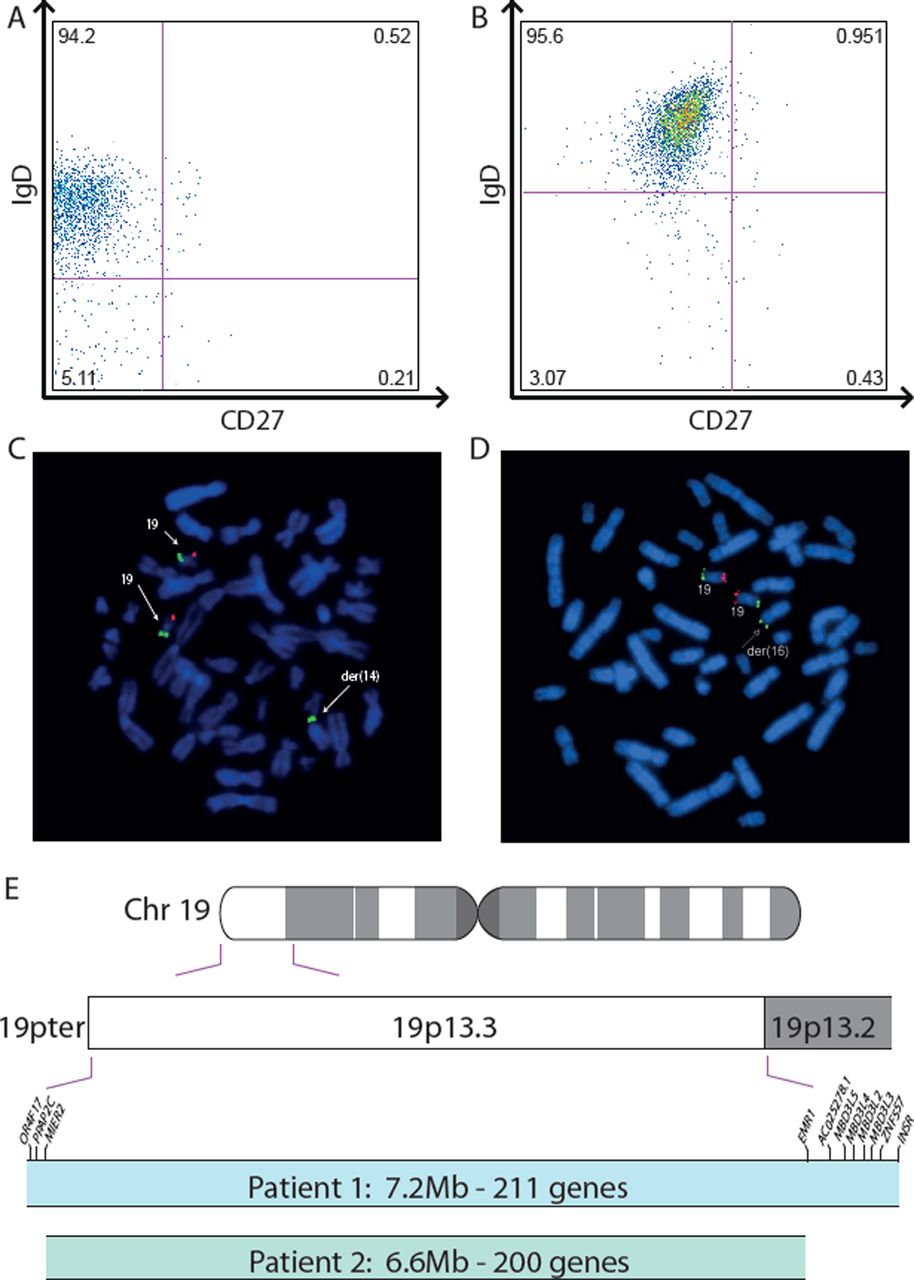
Both patients showed clear signs of humoral immunodeficiency in accordance with the clinical pattern of infections and presence of autoantibodies: in patient 1, global immunoglobulin levels and IgG subclasses were adequate for age (tables 2 and 3), but specific antibodies against pneumococcal antigens remained low after vaccination (table 3). Despite absence of clinical signs of autoimmunity, positive antinuclear and anticytoplasmatic autoantibodies (ANA and ANCA, resp.) were detected in patient 1 (table 2). Basic cellular immunological findings showed no overall quantitative alterations in T cell, B cell or natural killer (NK) cell counts. In order to further investigate the capacity of patients’ B cells to perform a class switch and develop memory responses, CD27 expression and IgD expression were investigated as described previously.19 ,20 This revealed that class switched B memory cells were reduced (table 4, figure 2A). In patient 2, humoral immunological abnormalities were more pronounced: overall IgG levels were mildly reduced with clear reduction of IgG1 and IgG3 subclasses and IgM, while IgA was normal (tables 1 and 3). Moreover, patient 2 had highly increased anti-double-stranded DNA antibodies (32 IU/mL; normal 0–15 IU/mL) and subclinical autoimmune thyroiditis on repeated occasions with high concentrations of thyroperoxidase antibodies (>1000 U/mL, normal: 0–60 U/L), thyroid stimulating hormone receptor antibodies (>100 U/L, normal: 0–15 U/L; table 4) and mildly increased thyroid stimulating hormone 5.89–8.6 µU/mL (normal 0.1–4.0 µU/mL) but normal thyroxin and only mildly elevated trijodthyronine. Additionally, 21-hydroxylase autoantibodies and glutamate decarboxylase autoantibodies were positive, suggesting imminent polyendocrinopathy as a potential sign of immune dysregulation in patient 2.
Laboratory abnormalities and treatment of two patients with partial trisomy 19 (19p13.3)
Humoral immune phenotype of two patients with partial trisomy 19 (19p13.3)
Cellular immune phenotype and autoantibodies of two patients with partial trisomy 19 (19p13.3)
{kind=link}
{kind=link}
Immunological and genetic phenotype. Despite normal B lymphocyte cell counts, IgD+CD27 and IgD-CD27 memory B cells were nearly absent in patient 1 (A) as well as in patient 2 (B). FISH analysis of patient 1 showed partial trisomy 19 with additional material from chromosome 19p on the short arm of chromosome 14 (C). FISH analysis of patient 2 revealed an unbalanced translocation from chromosome 19 to chromosome 16 (D). A schematic display of Chromsome 19 with an illustration of the duplicated regions in patients 1 and 2, respectively (E).
Cellular investigations revealed absolute numbers of T cells, B cells and NK cells to be normal in patient 2, however similar to patient 1, class switched B memory cells were substantially reduced (table 4 and figure 2B), suggesting a B cell maturation defect. While T cell proliferative responses were initially within normal range in both boys, recent data revealed a reduced T cell proliferation upon stimulation in vitro in the older patient 1. T cell receptor (TCR) excision circle analysis as a marker of thymic function showed no reduction, and T cell receptor Vβ repertoire to assess TCR rearrangement was unremarkable (table 4). NK cell cytotoxicity as determined by CD107a degranulation was borderline low in both patients, while cytotoxic T cell cytotoxicity was normal (table 4). Granulocyte and monocyte oxidative burst assays showed a moderate reduction (table 4). Complement activity was normal in both boys, but mannan-binding lectin (MBL) was practically absent in patient 2, yielding impaired lectin-associated complement activation. In patient 1, although basal MBL serum concentration was borderline normal, genotype analysis of MBL2 and its promoter showed polymorphisms that are usually associated with inadequate MBL production (table 3).
Other laboratory abnormalities
Additional laboratory investigations in patient 1 revealed hypovitaminosis D3 corresponding to clinical signs of osteopenia and latent hypothyreosis. Because patient 2 had mild anaemia with haemoglobin levels of 10 g/dL including microcytosis (mean corpuscular volume 69fl) and hypochromia (mean corpuscular haemoglobin 20pg; mean corpuscular haemoglobin concentration 29 g/dL) despite normal iron metabolism (ferritin and transferrin normal), and because his chromosomal aberration showed to involve the haemoglobin α chain locus on 16p, he was suspected to have α thalassaemia minor. Haemoglobinopathy workup revealed no clinically relevant haemoglobinopathy (normal Hb-electrophoresis, normal HbA, HbF, HbA2), but genetic data (see below) and the microcytic hypochromic anaemia were compatible with α thalassaemia trait (genotype –/αα).
Together, these findings (1) confirm and extend what is known on anatomical and neurodevelopmental abnormalities, and (2) newly describe a complex immunodeficiency phenotype involving mainly a memory B cell and humoral defect with increased risk of infections and features of immune dysregulation in patients with partial trisomy 19p13.
Genetic assessment
Results of CGH array, performed as described elsewhere,21 were annotated using Human Genome Build GRCh37/hg19. Genes within the break points were taken from http://www.ensembl.org22 and filtered for protein-coding genes only. CGH analysis of patient 1 revealed a 7.2 Mb terminal duplication of the short arm of chromosome 19 (arr(hg19) 19p13.3p13.2(90 897–7 300 043)×3). This region contains 211 known protein-coding genes (see online supplementary table S1). Moreover, FISH revealed a translocation from chromosome 19 to chromosome 14 (46,XY.ish der(14)t(14;19)(p11.2;p13.2)de novo) (figure 2C).
CGH analysis of patient 2 showed a 6.56 Mb terminal duplication of the short arm of chromosome 19 (see online supplementary table S1) combined with a partial 0.92 Mb deletion of Chromosome 16p13 including a total of 45 protein-encoding genes (see online supplementary table S2) ((arr(hg19) 16p13.3(106 271–1 024 153)×1,19p13.3(327 273–6 887 622)×3)). FISH revealed an unbalanced translocation from chromosome 19 to chromosome 16 (46,XY.ish der(16)t(16;19)(p13.3;p13.3)) (figure 2D). Parents of patient 2 and his older healthy sister did not consent to and thus did not undergo genetic analysis. The duplicated region of 19p13 consists of 200 protein-coding genes all of which were also contained in the duplicated region from patient 1 (figure 2E). By contrast, the duplication in patient 2 did not include several genes duplicated in patient 1, including OR4F17, PPAP2C, MIER2, EMR1, AC025278.1, MBD3L5, MBD3L4, MBD3L2, MBD3L3, ZNF557 and INSR.
Discussion
To date, few chromosomal disorders have been associated with primary immunodeficiency diseases.1 ,23 The best known is 22q11.2 deletion syndrome, including a DiGeorge phenotype and Velo-Cardio-Facial syndrome.1 ,2 Besides characteristic syndromic dysmorphies, those patients develop no or a hypoplastic thymus, leading to defective thymocyte development.2 The degree of immunodeficiency varies between the affected patients up to severe combined immunodeficiency with cellular and humoral defects.2 Although 22q11.2 deletion is the most frequent deletion syndrome (incidence 1:400024), and a defect of a T-box family transcriptional regulator, TBX1, relevant during organogenesis in mice and humans (reviewed in25) appears a candidate mechanism for 22q11.2 deletion syndrome, the genes that directly cause the involved immunodeficiency could not be identified so far2, which stresses the difficulty behind such intentions. Other microdeletion syndromes associated with impaired immunity include Wolf-Hirschhorn syndrome due to partial deletions of chromosome 4p1 ,3 and syndromes caused by deletion of chromosome 10p13-14 and partial deletions of chromosome 18.1 ,23 In addition, patients with 45,X0 phenotype (Turner's syndrome) may present with antibody deficiency and an increased predisposition to autoimmunity.1
Immunodeficiency associated with partial or complete chromosomal duplications is rare with the exception of for example, duplication of methyl-CpG binding protein 2 (MECP2), a regulator of gene expression and chromatin structure encoded on Xq28, which leads to a complex syndrome with mild dysmorphia, intellectual disability, neurological deterioration and variable degrees of immunodeficiency (including impaired gamma interferon (IFNg) secretion and T helper cell type 1 (TH1) responses, reduced numbers of memory T cells, B cells and NK cells in children) as confirmed in human and murine studies.26 Another chromosomal duplication syndrome that has long been associated with increased susceptibility to infections, especially severe respiratory tract infections, is trisomy 21. Those patients may show changes in thymus development and in B cell, T cell and NK cell counts, as well as low specific antibody response and granulocyte function.27
The present report is the first linking primary immunodeficiency to partial trisomy 19p13. The clinical and anatomical features of our patients are similar to reports on partial trisomy 19p13.3 described previously, including IUGR, neurological impairment (profound psychomotor retardation, microcephalus, epilepsy, sensorineural hearing loss), facial dysmorphy (flat nasal bridge, telecanthus, short palpebral fissures, low set ears, long philtrum) and urogenital malformations.4 ,6 ,7 ,9–11 ,13 The aforementioned publications often supplied only FISH mapping results (specific break points of the duplication were not examined), which makes it difficult to comparatively assess specific genes involved.4 ,6 ,7 ,9–12 More recently, Dolan et al8 described a patient with a microduplication within 19p13.12-19p13.2 supporting a list of involved genes; however, the genes duplicated in the patients being different. Furthermore, one patient with a smaller (0.81Mb) duplication within 19p13.3 was reported,13 sharing similar features with our patients, including IUGR, microcephalus, psychomotor development delay, seizures accompanied by high fever, hyperopia and some facial features (flat nasal bridge, posteriorly angulated ears). Among the 24 protein-encoding genes of this interval which are also duplicated in both of our patients, the authors highlight MAP2K2 and SEMA6B, as being potentially involved in the dysmorphia and psychomotor phenotype.13 MAP2K2 encodes for a mitogen-activated protein kinase, and loss of function of this gene product has been associated with Cardiofaciocutaneous syndrome 4 (OMIM #615280), a syndrome associating heart defects, facial dysmorphy and neurocognitive delay but no immunodeficiency. SEMA6B encodes semaphoring protein, which is involved in axon guidance and may contribute to the mental retardation observed in their patients.13
Intriguingly, previous publications lack laboratory results regarding the immune system. This is remarkable given that at least 2 of these 10 suffered from recurrent febrile episodes,10 ,12 1 of whom died in early childhood of respiratory syncytial virus pneumonia,12 and 1, additionally, had autoimmune disease.10
IgM deficiency may potentially contribute to the increased risk of upper and lower respiratory tract infections and autoimmunity as detected in combination with IgG subclass deficiency in patient 2. Although its relevance is uncertain as primary IgM deficiency may resolve spontaneously, it has been described in a number of syndromes with chromosomal abnormalities including microdeletion 22q11 (reviewed in28) and might represent another feature of impaired B cell maturation in partial trisomy 19p13.
Regarding the shared complex immune phenotype of our patients 1 and 2 with an increased frequency and severity of ear-nose-throat, upper and lower airway and invasive/systemic infections, compatible with the detected pathological immune phenotype of humoral and memory B cell deficiency, and moreover, reduced NK cell and phagocyte functions, the genes ELANE, TCF3, CD70 and C3 could be of particular interest. It remains speculative whether gene dosage might be involved in possible aberrant function of the respective gene products. ELANE, encoding neutrophil elastase, if mutated is known to cause severe congenital neutropenia,29 ,30 however the effect of a duplication of ELANE has not been investigated systematically. Although the described patients 1 and 2 have normal neutrophil counts, oxidative burst was moderately reduced in both cases.
TCF3, has been shown to play an important role in T cell and B cell differentiation31 and is involved in repeated chromosomal rearrangements connected with acute lymphoblastic leukaemia in children.32 Recently, a form of agammaglobulinaemia and absence of B cell antigen receptor was linked to deficiency of TCF3 (E2A),33 indicating that TCF3 might represent a candidate gene among the region of interest in the presented patients.
CD70 belongs to the tumour necrosis factor ligand family and binds to CD27 on B cells and T cells.34 CD70 is involved in cognate T–B interaction and plays a key role in promoting the differentiation of CD27 memory B cells into plasma cells.35 A primary immunodeficiency involving loss-of-function mutations in CD27 has recently been associated with common variable immunodeficiency like immunodeficiency and EBV-driven lymphoproliferation.20 ,36 Patients with common variable immunodeficiency show naïve B cells that cannot upregulate CD70, resulting in impaired class switch.34 ,37 TCF3 and CD70 might play a role in the pathogenesis in the patients described here, considering their reduced immunoglobulin levels and class-switched B memory cell numbers. Finally, among the duplicated genes, we find C3 encoding for complement factor 3, necessary in alternative and classical complement activation pathways.38 C3 deficiency is known to be associated with recurrent severe infections and in some cases with immune complex-mediated autoimmune diseases.39
In contrast to the over 6 Mb long duplication in both patients, the deleted region of patient 2 on 16p13.3 is much smaller (0.92 Mb) and contains only 45 protein-encoding genes, notably comprising the α haemoglobin locus. As has been reported previously, deletions within 16pter-p13.3 may lead to α-thalassaemia/mental retardation syndrome (ATR-16, OMIM #141750; 16pter-p13.3; for selected cases and review see 40–42). Patient 2 presents with normal Hb-electrophoresis, (normal HbA, HbF, HbA2) and mild microcytic hypochromic anaemia (Hb around 10 g/dL, mean corpuscular volume of 69fl, mean corpuscular haemoglobin of 20pg, mean corpuscular haemoglobin concentration of 29 g/dL) corresponding to an α thalassaemia trait (genotype --/αα). Nevertheless, patients with ATR-16 are described to show only mild mental impairment and minor anomalies, which also differ between affected individuals and are not fully consistent with the characteristics of our patients 1 and 2.41 ,42 Therefore we ascribe the outer appearance and mental retardation of patient 2 rather to the duplication of 19p13. This assumption is supported by the findings of Harteveld et al43, who focused on patients with ATR-16 without mental retardation and came to the conclusion that dysmorphic features and mental retardation are due to haploinsufficiency of an 800 kb region that does not overlap with the deletion of our patient 2.
As the immunodeficiency in patient 1 had been recognised before birth of patient 2, an early administration of Ig substitution was initiated in patient 2 and possibly explains improved thriving and may have protected him from a history of more severe infections.
Conclusion
Taken together, these data expand the known clinical spectrum of partial trisomy 19p13 to include primary immunodeficiency, and we suggest the acronym FURID19 (Facial dysmorphia, Urogenital malformation, growth and neurodevelopmental Retardation, ImmunoDeficiency, trisomy 19p13) for this syndrome. In general, immunodeficiency may be under-recognised in syndromic disorders, in particular in clinical situations where other features are prominent or implicate barrier dysfunctions. Thus, our findings underline that (1) patients with multiple malformations should undergo CGH array analysis and (2) patients with recurrent infections and syndromic features or microdeletions/microduplications should be tested for associated immunodeficiency to prevent a diagnostic and therapeutic delay and enable initiation of specific immunotherapy as early as possible.
Acknowledgments
The authors thank the patients and their families for understanding cooperation and giving their written informed consent to publish disease-specific data and features including photographs in the interest of advancing medical and scientific knowledge. Furthermore, fruitful discussions with Prof O A Haas and Prof M Speicher are highly appreciated and their scientific human genetic contribution to finalise the manuscript is acknowledged.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
MGS, CD, KB and EF-W contributed equally to this study.
-
Contributors All authors state that they fulfil all of the following criteria for authorship: (1) Substantial contribution(s) to conception and design, acquisition of data, or analysis and interpretation of data; (2) Drafting the article or revising it critically for important intellectual content; and (3) Final approval of the version to be published. In detail and in addition to above points (1)–(3), …authors state that:
MGS designed the study together with EF-W and KB, wrote the initial version and the final draft of the manuscript, drafted the figures and tables, acquired, analysed, and interpreted clinical, laboratory, immunological and genetics data of patient 2, recognised phenotypical features and is in charge of the clinical care of patient 2. CD drafted significant parts of the manuscript, recognised phenotypical features and is in charge of the clinical care of patient 1. SW acquired, analysed, interpreted phenotypical and laboratory data and interpreted human genetics analyses of patients 1 and 2 together with KB; additionally she drafted figures, tables and significant parts of the manuscript. ASN acquired, analysed and interpreted clinical neurological and laboratory, data, recognised phenotypical features and is in charge of the clinical care of patient 2 together with MGS.
ES-V performed experimental immunological assays in patient 1. KS interpreted clinical and immunological data, and is in charge of the clinical care of patient 1 together with EF-W and CD. JN acquired, analysed and interpreted clinical phenotypical and laboratory data, recognised phenotypical features and is in charge of human cytogenetics analyses of patient 1. SU acquired, analysed and interpreted clinical phenotypical and laboratory data, recognised phenotypical features and is in charge of human genetics analyses of patient 2. WFP generated and analysed immunological laboratory data. WS acquired, analysed and interpreted clinical, laboratory, and substantial amounts of immunological data of patients 1and 2, and is in charge of the clinical care of patient 2. CU interpreted clinical, haematological laboratory, and substantial amounts of immunological data, and is in charge of the clinical care of patient 2. KB designed the study, acquired, analysed and interpreted immunological and genetics data of patients 1and 2, wrote the first draft and the revised version of the manuscript together with MGS, CD and EF-W and revised the final version of the manuscript. EF-W designed the study, interpreted clinical, laboratory, immunological and genetics data, recognised phenotypical features and is in charge of the clinical care of patient 1, and revised the final version of the manuscript.
-
Funding This work was supported by intramural funds of the CeMM Research Institute for Molecular Medicine of the Austrian Academy of Sciencies and the FWF START Programme (both to KB), and an intramural fund of the Dept. Paediatrics and Adolescent Medicine Vienna supports the work of CD and EF-W.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Medical University Vienna Ethics Commission, Approval No. 499/2011 for immunologic and genetic characterization of pediatric patients with immunodeficiencies.
-
Provenance and peer review Not commissioned; externally peer reviewed.