Article Text

Abstract
Background Distinguishing genetic variants that cause disease from variants that are rare but benign is one of the principal challenges in contemporary clinical genetics, particularly as variants are identified at a pace exceeding the capacity of researchers to characterise them functionally.
Methods We previously developed a novel method, called paralogue annotation, which accurately and specifically identifies disease-causing missense variants by transferring disease-causing annotations across families of related proteins. Here we refine our approach, and apply it to novel variants found in 2266 patients across two large cohorts with inherited sudden death syndromes, namely catecholaminergic polymorphic ventricular tachycardia (CPVT) or Brugada syndrome (BrS).
Results Over one third of the novel non-synonymous variants found in these studies, which would otherwise be reported in a clinical diagnostics setting as ‘variants of unknown significance’, are categorised by our method as likely disease causing (positive predictive value 98.7%). This identified more than 500 new disease loci for BrS and CPVT.
Conclusions Our methodology is widely transferable across all human disease genes, with an estimated 150 000 potentially informative annotations in more than 1800 genes. We have developed a web resource that allows researchers and clinicians to annotate variants found in individuals with inherited arrhythmias, comprising a referenced compendium of known missense variants in these genes together with a user-friendly implementation of our approach. This tool will facilitate the interpretation of many novel variants that might otherwise remain unclassified.
- Genetics
- Cardiovascular Medicine
- Clinical Genetics
- Congenital Heart Disease
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Inherited arrhythmias such as long QT syndrome (LQTS), Brugada syndrome (BrS) and catecholaminergic polymorphic ventricular tachycardia (CPVT) are life-threatening diseases, caused predominantly by genetic variation in ion channels. In BrS, loss-of-function mutations in the SCN5A-encoded cardiac sodium channel (MIM 601144) have been shown to be responsible for 15–30% of cases1 ,2 with mutations in other minor genes accounting for a proportion of remaining cases. In CPVT, gain-of-function mutations in the cardiac ryanodine receptor encoded by RYR2 (MIM 604772) are responsible for at least 50% of cases.3
With on-going developments in DNA sequencing technology, it is expected that clinical genetic testing will become very widely available. However, it is increasingly appreciated that many healthy individuals carry rare variants in disease-associated genes (3–6% for genes associated with inherited arrhythmia syndromes),4 ,5 and that additional information is required to determine whether a novel variant identified during genetic testing is pathogenic. This is particularly the case for missense variants (single amino acid substitutions) caused by single nucleotide variants.
In order to determine whether a rare variant found in a patient is disease causing, sufficiently powered segregation analysis or functional biochemical characterisation are ideally performed. However, these are often impractical due to cost and time constraints, or a lack of phenotypically characterised family members for segregation studies. Several in silico algorithms, such as SIFT and Polyphen6 ,7 try to predict the effect of novel variants, based on the conservation and physicochemical properties of the variant amino acid, and variants in certain protein regions and domains have a high probability of pathogenicity.8 ,9 However, more evidence is needed to classify variants with sufficient confidence for clinical application.
We have recently proposed a new method to analyse novel missense variants and assess their likelihood of pathogenicity.10 This method, ‘paralogue annotation’, identifies functionally important residues that are intolerant of variation across families of evolutionarily related proteins (paralogues), using clinically ascertained human genotype–phenotype relationships. By aligning the protein sequences of members of these protein families, we can identify amino acids that are functionally equivalent across the different proteins. A variant that is known to be pathogenic in one member of a protein family can then be used to annotate the equivalent amino acid of other members of the family for which no clinical genetic information exists (figure 1). For example, if a missense variant in RYR1 alters protein function and causes malignant hyperthermia when expressed in skeletal muscle, then we hypothesise that a novel variant affecting the equivalent amino acid in RYR2, expressed in cardiac muscle, is likely to be disease causing in a patient with CPVT.
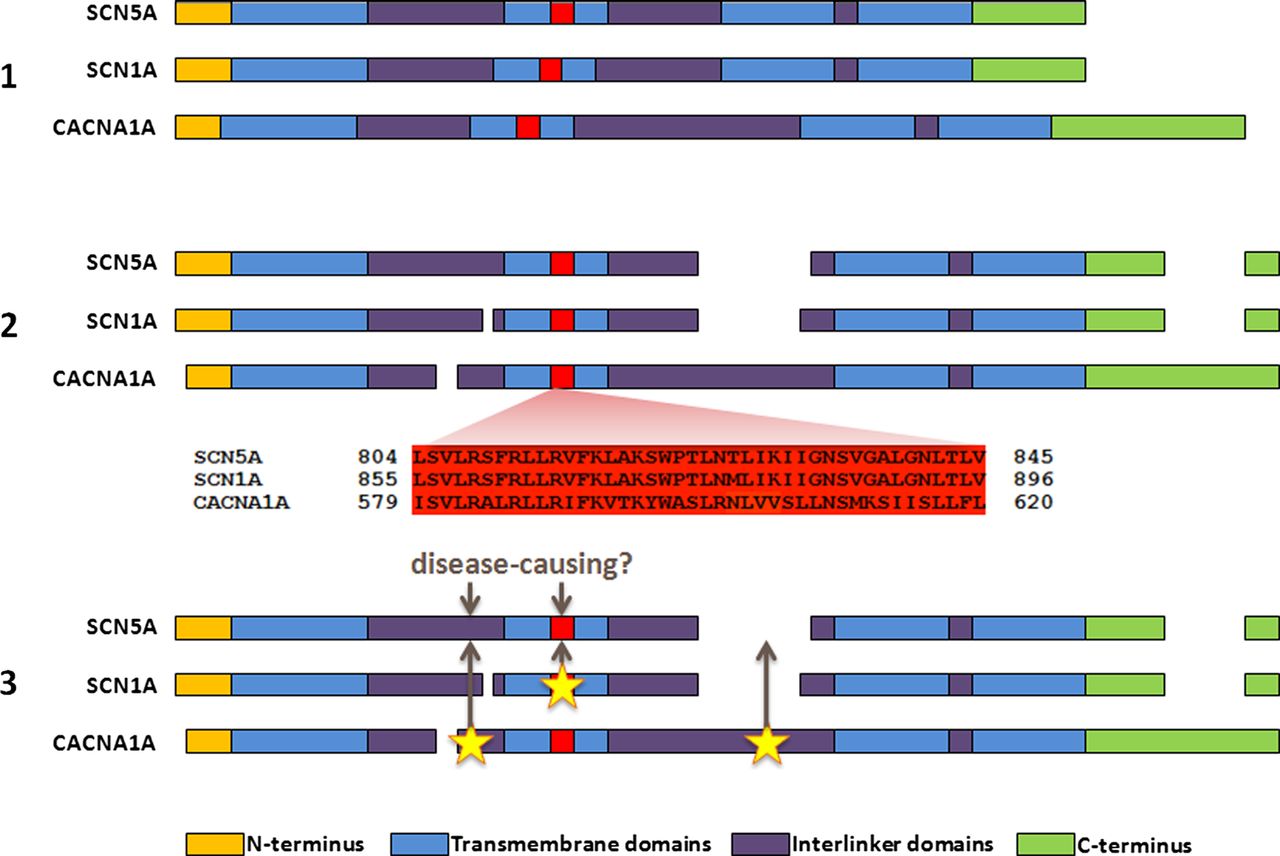
An overview of paralogue annotation. (1) Paralogues (evolutionarily related genes, with homologous sequence and protein domain structures) are identified for a gene of interest. A subset of paralogues for SCN5A is shown here for illustration. (2) Protein sequences of paralogues are aligned, identifying functionally equivalent amino acids across the protein family. (3) Disease-causing variants in paralogues are identified from previous literature reports, and their locations are mapped to the gene of interest. Variants at these sites have a high probability of pathogenicity.
This approach was developed and experimentally validated by application to a large set of known variants in eight LQTS genes, and was found to have a positive predictive value (PPV) of 98.4% in these genes.10 Here we present novel refinements to increase accuracy, and apply this approach in two large cohorts of patients with BrS or CPVT to determine whether it provides additional useful information in a clinical diagnostic setting. We also report a web application that allows researchers and clinicians to use paralogue annotation to interrogate novel variants in arrhythmia genes.
Materials and methods
Identification of variants in paralogues of RYR2 and SCN5A
Paralogues of RYR2 and SCN5A, that is, evolutionarily related genes with homology in sequence and structure, were identified using the IUPHAR database on receptor nomenclature11 and through homology searches (Blastp searches of the SCN5A and RYR2 protein sequences against the human Refseq protein database).12 The transcripts and protein isoforms used for these genes were RYR2: NM_001035/NP_001026 (Refseq), ENST00000366574/ENSP00000355533 (Ensembl), LRG_402t1/LRG_402p1 (Locus Reference Genomic) and SCN5A: NM_198056/NP_932173 (Refseq), ENST00000333535/ENSP00000328968 (Ensembl), LRG_289t1/LRG_289p1 (Locus Reference Genomic).
RYR2 has two paralogues, RYR1 and RYR3, which were included in the sequence alignment. Although RYR3 is not implicated in human disease it was included to improve the alignment quality for the protein family. Variants in RYR1 cause malignant hyperthermia, central core disease, multi-minicore disease and congenital myopathy. There are 329 RYR1 missense variants in the Human Gene Mutation Database (HGMD) affecting 284 distinct amino acid residues.
For SCN5A, 19 paralogues were included: nine voltage-gated sodium channels, and 10 voltage-gated calcium channels that show strong sequence and structural homology with the sodium channels. Fourteen of these 19 paralogues are associated with Mendelian disease in humans, typically attributable to altered cellular electrophysiology in the tissue where the paralogue is expressed, such as epilepsy, myotonia, pain disorders, night blindness and hemiplegic migraine. SCN5A paralogues and their disease associations are shown in table 1.
SCN5A paralogues
For each paralogue gene, variants reported as pathogenic were identified using the HGMD Professional V.2012.3.13 Only disease-causing missense mutations, that is, single nucleotide variants causing a single non-terminal amino acid change, were considered.
Multiple sequence alignment and paralogue annotation
The protein sequences of RYR2 and SCN5A and their respective paralogues were aligned using the T-Coffee/M-Coffee alignment packages.14 These packages combine the output of a number of alignment algorithms into a single consensus alignment and provide a consensus score at each point in the alignment (0–9), which is a measure of the reliability of the alignment at each amino acid residue. Using these alignments, each paralogue protein residue with a disease-causing variant was mapped onto the equivalent amino acid residue in RYR2 and SCN5A.
To distinguish aligned amino acids that are truly functionally equivalent from alignment artefacts, an amino acid alignment quality classification was devised. Mappings were classified as high quality if the reference amino acid was conserved between paralogues, and the alignment consensus score was greater than 3. Medium quality mappings required conservation of the reference amino acid or a consensus score greater than 3 or more than one paralogue variant mapping to the same residue. Low quality mappings did not meet these criteria, and were excluded from analyses.
Sequencing and paralogue annotation in cohorts with CPVT and BrS
Sequencing of two large cohorts of unrelated patients with inherited arrhythmias has previously been reported: all exons of RYR2 were sequenced in 155 patients with CPVT and 200 healthy controls,4 and all exons of SCN5A were sequenced in 2111 patients with BrS and 1300 healthy controls.5 Paralogue annotation was applied to novel missense variants identified in each cohort. Variants were classified as ‘novel’ if they had not previously been reported at the time of the original report, and have not subsequently been confirmed in an independent study. These would conventionally be reported clinically as ‘variants of unknown significance’, pending segregation and/or functional molecular characterisation.
Mutation status of RYR2 and SCN5A residues
Previous reports of benign and pathogenic variation in RYR2 and SCN5A were collated. HGMD identified putative disease-causing variants in both genes. Variants were also extracted from dbSNP V.137 as part of the Ensembl 69 release. Population frequency from 1000 Genomes (phase 1) and other datasets in dbSNP were also used—any variant with more than one observation and a minor allele frequency (MAF) of greater than or equal to 0.01 in any population was classified as benign, as this is considered incompatible with the population frequencies of BrS and CPVT. In addition, several large published datasets of RYR2 and SCN5A pathogenic and benign variants were assessed.15–20
Residues with one or more missense variants reported as disease causing were classified as pathogenic. Residues with reported benign missense variants or high MAF scores as described above were classified benign. Any residue with conflicting benign and pathogenic reports (for the same variant or different variants affecting the same residue), or dbSNP variants with no population frequency data or MAF of less than 0.01, were classified as uncertain. Residues for which no missense variants were known were classified as unannotated.
Protein domains of RYR2 and SCN5A
Mutation hotspots in RYR2 were defined according to Yano et al:9 residues 77–466 (N-terminal hotspot), 2246–2534 (central hotspot) and 3778–4959 (channel hotspot). The SCN5A domains were defined in accordance with the Uniprot entry Q14524 as used by Kapa et al:8 transmembrane regions are between residues 127–415, 712–939, 1201–1470 and 1524–1772.
Results
Paralogue annotation is informative in assessing novel missense variants in clinical cohorts
Variants identified in two large cohorts of unrelated patients with inherited arrhythmias were analysed. RYR2 sequencing in 155 CPVT patients identified 63 distinct variants that were absent in 200 healthy control references. Of these, 59 were missense variants, and 31 were not reported in any other previous or subsequent cases. SCN5A sequencing 2111 BrS patients identified 293 distinct SCN5A variants that were absent in 1300 healthy controls. Of these, 193 were missense variants, and 122 were not reported in any other previous or subsequent cases. Paralogue annotation was applied to these 31 RYR2 and 122 SCN5A variants, which would otherwise be reported clinically as ‘variants of unknown significance’, to determine whether paralogue mutation annotation is informative and provides further evidence to reclassify some of these variants as ‘disease causing’ (figure 2).

Genetic variation in cohorts with inherited arrhythmia syndromes. RYR2 or SCN5A were sequenced in 2266 patients with an inherited arrhythmia syndrome, as shown. Three hundred and fifty-seven variants were identified in 511 patients, of which 153 were novel missense variants. In the absence of segregation or functional data, these would typically be reported as variants of unknown significance. However, paralogue annotation provided additional information for 65 (36%) variants, that would further inform a clinical genetic report. BrS, Brugada syndrome; CPVT, catecholaminergic polymorphic ventricular tachycardia.
Ten of the 31 novel RYR2 missense variants (32.3%) affected amino acid residues that were annotated with paralogue mutations, that is, variants in the equivalent residue in RYR1 have been shown to be disease causing for other human diseases (table 2). Five of these 10 residues have more than one reported RYR1 pathogenic mutation. Three of these variants occurred in the N-terminal hotspot (residues 77–466), five occurred in the channel region hotspot (residues 3778–4959) and two occurred outside the recognised RYR2 mutation hotspots—A549 V and H2168Q. In addition, five of the 29 non-novel RYR2 missense variants identified in the study also had equivalent paralogue mutations, adding further evidence to the likely pathogenicity of these variants (see supplementary table S1, available online only).
Paralogue annotation of novel variants identified in RYR2
In SCN5A, 45 of the 122 novel missense variants (36.9%) were annotated with paralogue mutations (table 3). Fourteen of these 45 residues have more than one reported paralogue pathogenic mutation. Forty-one of these variants occurred in the transmembrane domains of SCN5A, three in the N-terminal region and one in the interlinker domains. In addition, 26 of the 70 non-novel SCN5A missense variants identified in the study also had equivalent paralogue mutations, adding further evidence to the likely pathogenicity of these variants (see supplementary table S2, available online only).
Paralogue annotation of novel variants identified in SCN5A
Three variants that are detected in healthy controls in these two studies are predicted to be pathogenic using our approach, and are considered false positives for the purpose of this study. RYR2-M2389 L was identified in one of 200 controls. This variant is found in the central domain hotspot of RYR2 and is not identified in any of the 1000 Genomes phase 1 samples. SCN5A-D596G was identified in one of 1300 controls. Located in interlinker domain I–II, this variant is also absent in the phase 1 1000 Genomes data. SCN5A-S216 L has conflicting reports as to its pathogenicity—it has been linked to LQTS, atrial fibrillation and dilated cardiomyopathy,21–23 but has also been identified in healthy controls8 and in low frequencies in dbSNP populations. Its exact role in causing disease is therefore still unclear. Although we consider these variants as false positives here, in the absence of functional data to the contrary, at least some may be functionally deleterious in controls with non-penetrant disease or incomplete phenotype data. Importantly, controls did not undergo any provocation testing (eg, ajmaline challenge for BrS, and exercise test or adrenaline challenge for CPVT), which is used to make a diagnosis in some forms of these diseases and hence latent disease was not excluded.
Paralogue annotation has previously been validated in eight LQTS proteins.10 Sites predicted to be disease associated by paralogue annotation were highly enriched for known disease-causing variants, with very few false positives. Applying the approach to RYR2 and SCN5A variants again demonstrates enrichment for known disease-associated residues (table 4). Two hundred and seventy-five RYR2 residues were annotated with paralogue mutations: if distributed at random, eight would be expected to coincide with known pathogenic residues. We observe 35 known disease-associated residues: a 4.4-fold enrichment. Similarly, 113 disease-associated SCN5A residues are annotated by paralogue mutations, a 1.4-fold increase over the 81 annotations expected by random distribution. There is also a significant depletion of known benign residues in SCN5A (expected, six; observed, one). The positive predictive value (PPV) of paralogue annotation assessed in the current study is 98.7%, in keeping with previous findings in LQTS genes (98.4% PPV).10 By comparison, of 519 known missense variants in RYR2 and SCN5A predicted by SIFT to be deleterious, 500 are correct (variants with unambiguous evidence of pathogenicity) with 19 false positives (benign variants), yielding a PPV of 96.5%. Similarly, of 518 variants predicted to be damaging by Polyphen, 497 are correct with 21 false positives, with a PPV of 95.9%.
Paralogue annotation accurately identifies disease-associated residues in RYR2 and SCN5A
Importantly, the paralogue approach is informative for a large number of residues at which no variation has previously been reported, when variation detected in clinical testing would otherwise be reported as a ‘variant of unknown significance’. Paralogue annotation is informative for 238 unannotated RYR2 residues and 321 unannotated SCN5A residues, and was informative for more than one-third of novel variants identified in these large clinical cohorts. It is expected that any missense variants identified in these residues in CPVT or BrS patients are likely to be disease causing.
Distribution of variation across protein domains
Protein regions that are known to be enriched for pathogenic variation were also enriched in paralogue annotations (compared with expectation under a random distribution; table 5). One hundred and seventy-two of the 275 paralogue mutations mapped to RYR2 occur in the three mutations hotspots, an enrichment factor of 1.52. Similarly, 227 of the 441 paralogue mutations mapped to SCN5A occur in the four transmembrane domains, an enrichment factor of 1.57. As might be expected, disease-causing variants display similar domain distributions across the protein families: RYR1 has similar overlapping mutation hotspots to RYR2 (figure 3) and 75.8% of the missense mutations in the voltage-gated sodium and calcium channel paralogues of SCN5A occur in their transmembrane domains, despite these regions only accounting for 50.5% of the protein length.
Annotation across protein domains of RYR2 and SCN5A

Disease-causing variation in human ryanodine receptors, RYR1 and RYR2. An alignment of RYR1 and RYR2 reveals the structural similarity of the proteins and homologous clustering of pathogenic variation in these related proteins. The protein is represented in light grey, with reported ‘mutation hotspots’ marked in dark grey, and exon boundaries highlighted. The locations of missense variants previously reported to be pathogenic are shown with black lines above and below the protein graphic—longer lines indicate more than one pathogenic DNA variant affecting the same protein residue.
Reciprocal paralogue annotation: RYR1
This technique is also applicable in a reciprocal fashion, that is, arrhythmia-causing mutations in RYR2 and SCN5A can be used to interpret novel variants found in any of the paralogues analysed in this study. As an example, we have mapped all the mutations in RYR2 that cause CPVT and other cardiac diseases onto RYR1, which can be used to assess novel missense variants found in patients with malignant hyperthermia or central core disease.
As expected, residues predicted to be intolerant of variation by paralogue annotation are indeed highly enriched for known disease-associated variants in RYR1 (4.4-fold enrichment; observed, 35; expected, eight). None of the four RYR1 residues that host missense variants with a MAF greater than 0.01 in 1000 Genomes (that are therefore presumably benign) were annotated, giving a notional PPV of 100%. In addition, 99 previously unannotated RYR1 residues are predicted to be intolerant of variation, of which 22 lie outside the traditional RYR1 mutation hotspots.
Web-based paralogue annotation application
We have developed a web-based tool to allow researchers and clinicians to identify paralogue annotations for novel variants to inform pathogenicity. This is available for RYR2 and SCN5A, as well as other LQTS-associated genes previously reported. Users input the coordinates of a novel variant (protein or complementary DNA coordinates) in a gene of interest, and any paralogue mutations associated with that residue are returned, together with fully referenced details of any known (pathogenic or benign) missense variants in the query gene. The quality of the mapping is crucial when using paralogue annotation to predict pathogenicity. The web interface will assign each mapping a high, medium or low quality rating (defined using the alignment consensus, reference amino acids in the query and paralogue proteins and number of mappings as described in the Methods section above). In addition, the segment of the multiple sequence alignment around the mapping is displayed to allow users to assess the degree of homology visually between the proteins at that residue, coloured by the consensus between the different alignment algorithms used by the M-Coffee and T-Coffee packages. Finally, links to the publications describing the pathogenic paralogue mutations are available, allowing users to assess how well the mutation has been characterised in the paralogue-associated disease. The application is shown in figure 4.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
A web-based application makes paralogue annotation easily accessible for genes causing inherited arrhythmia syndromes. A web-based application is available at http://cardiodb.org/Paralogue_Annotation/. Users enter the position of a novel variant using complementary DNA or protein coordinates: in this example a substitution has been found in RYR2, affecting glycine residue 357.1 This residue maps to RYR1 residue 341, and two cDNA variants at that location (c.1021G>A and c.1021G>C) that each cause substitution of Arg for Gly at that position have been reported to cause malignant hyperthermia. Users should check the alignment quality—here the mapping quality is high: the surrounding region is highly homologous, the reference amino acid is the same in both proteins, and the alignment has a high consensus score.2 Pubmed links give access to the reports relating to the paralogue mutation(s), 3 allowing users to assess the quality of evidence for pathogenicity. Here functional characterisation has been performed on this variant in the highlighted publication, adding confidence that the variant is disease causing and the residue is intolerant of variation in both RYR1 and its paralogue RYR2.
Discussion
In this study we have shown that over one-third (36%) of novel missense variants identified in large cohorts of patients with CPVT (RYR2) and BrS (SCN5A) can be annotated by paralogue mutations. This additional evidence of pathogenicity may allow us to reclassify as disease causing up to one-third of variants currently reported as ‘variants of unknown significance’, with a positive PPV exceeding 98%.10 There is a bias towards reporting pathogenic variants over benign variants in the literature, and although we have supplemented literature reports with data from 1000 Genomes and the Exome Sequencing Project, it is possible that these PPV values may be overestimated. However, as these data derive from clinically observed genotype–phenotype relationships within closely related protein families, rather than theoretical predictions based on the sequence and structure of much more distantly related proteins, we have confidence in this approach and these annotations.
In a clinical setting, the identification of a novel single amino acid substitution in a gene associated with inherited disease is generally insufficient to report the variant as disease causing. This is due to the relatively high prevalence of rare, benign variants in healthy controls in many genes. Missense variants have been shown to be present in 6% of controls in RYR24 and in 2.7% of controls in SCN5A.8 Segregation studies and functional biochemical characterisation are powerful, but resource intensive and are not always applicable. Computational predictions are attractive and widely applicable, but while informative they are not sufficiently accurate for robust clinical reporting. It therefore behoves us to make full use of available reports linking human variation to clinical phenotype.
When a variant in a disease gene has previously been observed, either in association with disease or in apparently healthy individuals, this provides invaluable additional information for clinical reporting. However, there is a huge amount of additional data linking genetic variation to clinical phenotype in paralogues of disease genes that is not presently used. By mapping functionally equivalent amino acid residues across protein families, and transferring genotype–phenotype annotations between related proteins, we can leverage this wealth of existing data to aid our interpretation of the significance of novel variants in a clinical setting.
The accuracy of this method depends on the quality of the protein sequence alignment (to ensure that aligned amino acids can be confidently regarded as functionally equivalent) and the quality of the evidence relating genotype to phenotype for the paralogue variant. For the alignment, appropriate paralogues should be carefully and manually chosen, while we have described methods above to ensure that only mappings of sufficient quality are noted. In addition, our web application allows users to assess the quality of the alignment visually at the mapping residue. The web application also provides Pubmed links to allow researchers to assess the quality of the reports linking the paralogue variants to disease—a functionally characterised variant, or one that has been shown to segregate with disease, is more informative than a variant simply observed once in an affected individual. It is critical for researchers to consider this evidence carefully to avoid inferring pathogenicity from erroneous reports and also check that the direction of effect of the variant in the paralogue is compatible with the observed phenotype in the gene of interest.
The sensitivity of this approach depends on the availability of known, pathogenic mutations in the paralogues of genes of interest. The lack of paralogue annotation for a novel variant does not imply that the variant is non-pathogenic, simply that a disease-causing mutation in the equivalent paralogue residue has not yet been observed. As genetic testing becomes more widely available across the whole spectrum of inherited disease, for example, through the ongoing development of sequencing technologies, we expect the number of informative paralogue mutations to expand greatly, increasing the sensitivity of this method. Here, an additional 238 residues in RYR2 (a 171% increase compared with previous reports), and 321 residues in SCN5A (87% increase) are putatively annotated as intolerant of variation.
Paralogue annotation is designed to interpret individual variants in monogenic disease and does not currently help to clarify the genetics of complex multi-alleleic disease. There have been recent reports linking common genetic variation with predisposition to BrS,24 and there is a debate about the role of genetic testing in this condition. However as BrS does segregate as a monogenic Mendelian trait in some pedigrees and most clinicians still feel that genetic testing of SCN5A is of value, paralogue annotation can help to interpret these novel variants.
Some regions of RYR2 and SCN5A are enriched for disease-associated variants, yet contain relatively few benign polymorphisms, so that variants in these regions have a higher-than-average previous probability of pathogenicity. While this is to some extent informative, the accuracy and sensitivity of a purely domain-based prediction are limited, as many pathogenic and benign variants do not follow this trend. Paralogue annotation provides a higher resolution annotation, down to the level of the individual amino acid. It is also not limited to any specific domains or protein regions and is less likely to mis-annotate a rare benign variant as pathogenic given the high PPV scores we have seen in this and previous studies.
In RYR2, 134 of the 154 known pathogenic missense mutations occur within the recognised mutation hotspots (figure 3). However, 10 known benign missense variants also occur in these regions, suggesting that novel variants found here cannot be definitively classified as disease causing. In our study we found that 57% of the paralogue annotations of RYR2 occur in the mutation hotspots. This is due to the fact that RYR1 contains equivalent, and largely overlapping, mutation hotspots (figure 3). Pathogenic mutations in RYR1 are, however, less concentrated in these hotspots than in RYR2, which allows us to annotate an additional 114 residues outside conventional RYR2 hotspots. It is likely that this clustering of mutations is at least partly caused by sequencing bias, that is, often only exons with known mutations are sequenced in genetic testing. The increasing use of next generation sequencing technologies allows laboratories to sequence all the exons of ryanodine receptors and is likely to expand the numbers of mutations found outside the hotspots of both RYR1 and RYR2 for their respective diseases—paralogue annotation will be able to utilise all of these findings to assess variants in either gene.
Pathogenic mutations are not as discreetly clustered in SCN5A as in RYR2. However, variation in the transmembrane regions is rare in apparently healthy individuals, and substitutions in these regions have been reported to have an 88% probability of pathogenicity when found in clinically affected individuals.8 Here, over 80% of disease-causing variants in SCN5A paralogues map to transmembrane residues, providing further evidence that these domains are functionally important and intolerant of variation. Most importantly, paralogue annotation also annotates some variants at residues outside these regions, whose significance would otherwise be unappreciated.
Paralogue annotation of variants is widely applicable: approximately half of all disease-associated genes have one or more paralogues with disease-causing variants.10 The technique can also be applied in a reciprocal manner, as illustrated here with RYR1 and RYR2, and our web-based application will be informative for researchers and clinicians interested in a range of diseases mediated by ion channels and membrane excitability, such as malignant hyperthermia and central core disease, and as well as the cardiovascular genetics community.
Conclusion
This study demonstrates an accurate and widely applicable approach to interpret novel missense variants, here applied to annotate variants in CPVT (RYR2) and BrS (SCN5A). This is informative for more than one-third of novel variants in these genes, and may provide sufficient evidence to report these variants as disease causing, rather than as ‘variants of unknown significance’, with a positive PPV of 98.7%. This approach is based on clinical genotype–phenotype relationships in humans, rather than computational prediction, giving further confidence in its application. While this method will not be applied in isolation, due to our incomplete inventory of pathogenic variation in paralogues, it provides an invaluable additional tool for clinicians and researchers. Our web-based application provides a user-friendly implementation of this technique for immediate application in the interpretation of novel variants, with an initial focus on inherited arrhythmia syndrome genes, together with an annotated compendium of previously reported missense variants in these genes.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online tables
Footnotes
-
Contributors RW and JSW devised the study. RW analysed the data and drafted the paper. JSW, SAC and NSP reviewed the data and revised the manuscript.
-
Funding This research was supported by the Academy of Medical Sciences, the Wellcome Trust, the British Heart Foundation (grant number SP/10/10/28431), Arthritis Research UK, Fondation Leducq and the NIHR Cardiovascular Biomedical Research Unit at Royal Brompton & Harefield NHS Foundation Trust and Imperial College London.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.