Article Text

Abstract
Background Jeune asphyxiating thoracic dystrophy (JATD) is a rare, often lethal, recessively inherited chondrodysplasia characterised by shortened ribs and long bones, sometimes accompanied by polydactyly, and renal, liver and retinal disease. Mutations in intraflagellar transport (IFT) genes cause JATD, including the IFT dynein-2 motor subunit gene DYNC2H1. Genetic heterogeneity and the large DYNC2H1 gene size have hindered JATD genetic diagnosis.
Aims and methods To determine the contribution to JATD we screened DYNC2H1 in 71 JATD patients JATD patients combining SNP mapping, Sanger sequencing and exome sequencing.
Results and conclusions We detected 34 DYNC2H1 mutations in 29/71 (41%) patients from 19/57 families (33%), showing it as a major cause of JATD especially in Northern European patients. This included 13 early protein termination mutations (nonsense/frameshift, deletion, splice site) but no patients carried these in combination, suggesting the human phenotype is at least partly hypomorphic. In addition, 21 missense mutations were distributed across DYNC2H1 and these showed some clustering to functional domains, especially the ATP motor domain. DYNC2H1 patients largely lacked significant extra-skeletal involvement, demonstrating an important genotype–phenotype correlation in JATD. Significant variability exists in the course and severity of the thoracic phenotype, both between affected siblings with identical DYNC2H1 alleles and among individuals with different alleles, which suggests the DYNC2H1 phenotype might be subject to modifier alleles, non-genetic or epigenetic factors. Assessment of fibroblasts from patients showed accumulation of anterograde IFT proteins in the ciliary tips, confirming defects similar to patients with other retrograde IFT machinery mutations, which may be of undervalued potential for diagnostic purposes.
- Clinical Genetics
- Molecular Genetics
- Developmental
- Diagnostics
- Genetic Screening/Counselling
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
Asphyxiating thoracic dystrophy or Jeune syndrome (Jeune asphyxiating thoracic dystrophy (JATD); MIM 208500), first described in 1955,1 is a rare genetically heterogeneous disorder characterised by skeletal anomalies, primarily shortened ribs and limbs, brachydactyly and polydactyly. Constriction of the thoracic cage is associated with recurrent respiratory infections in particular in neonates and infants, and in 60% of cases with lethal respiratory distress.2 Up to 30% of JATD patients also develop end stage renal disease,3 with hepatic fibrosis and retinal involvement reported less frequently.4 ,5 JATD is a member of the family of skeletal ‘ciliopathies’, disorders associated with dysfunction of primary cilia, classified as one of the six short-rib polydactyly syndrome (SRPS) disorders.6 ,7 JATD along with Ellis-van Creveld syndrome is an SRPS compatible with life, rather than one of the four lethal SRPS subtypes (SRPS I–IV).7 In addition, JATD is both phenotypically as well as genetically related to Sensenbrenner syndrome (Cranioectodermal dysplasia; MIM 218330)8 and Mainzer-Saldino syndrome (Conorenal syndrome; MIM 266920).9
Mutations in several different genes cause JATD. These genes, IFT80,10 TTC21B/IFT139,11 IFT140,9 ,12 WDR19/IFT1448 and DYNC2H113 all encode proteins that participate in ciliary intraflagellar transport (IFT), an evolutionarily conserved process which is essential for ciliogenesis and governs a variety of important cell signalling events that are key to normal human development.14 ,15 In IFT, two protein complexes IFT-A and IFT-B are bidirectionally transported in cilia by molecular motors, along with essential ciliary cargos.16 ,17 The IFT80 protein is part of the IFT-B particle that consists of at least 14 proteins, which in association with kinesin-2 motors drives anterograde transport from the ciliary base to the tip while IFT139, IFT140 and IFT144 are all part of the IFT-A particle that consists of six proteins and in association with the cytoplasmic IFT dynein-2/1b motor drives retrograde transport from the ciliary tip to the base.17–19 The other three IFT-A members, IFT122, IFT43 and WDR35, are mutated in Sensenbrenner syndrome.20–22 Similarly, IFT144 and IFT80 are also mutated in overlapping milder syndromes and in more severe type III short-rib polydactyly.8 ,23 IFT140 mutations were recently described in Mainzer-Saldino syndrome patients, and a subset of JATD patients with early end stage renal disease and frequent retinopathy.9 ,12 Besides DYNC2H1 deficiency underlying JATD, 13 mutations have also been reported in patients with two of the severe short-rib polydactyly subtypes, Majewski syndrome (SRPS type II; MIM 263520)24 ,25 and Verma-Neumoff syndrome (SRPS type III; MIM 263510).13 ,26 One SRPS type II family carrying double heterozygous pathogenic mutations in both NEK1 and DYNC2H1 was also suggestive of possible digenic inheritance.24 ,25 NEK1 dysfunction affects ciliogenesis and since the protein was localised to the basal body and within the cilium, a role in IFT was suggested.24 ,25
The DYNC2H1 protein, initially identified in sea urchin embryos,27 is the central ATPase subunit of IFT dynein-2 complex, the principle minus-end directed microtubule motor that drives retrograde transport of the IFT-A particle to regulate tip-to-base transport.15 ,28 ,29 DYNC2H1 has a typical dynein heavy chain organisation (figure 1) similar to other heavy chain dyneins underlying primary ciliary dyskinesia and other developmental disorders.34–39 DYNC2H1 knockdown in Caenorhabditis elegans and mice results in short cilia with bulged tips,40 ,41 and Hedgehog signalling, an important signalling pathway that regulates bone development, is disrupted in Dync2h1-deficient mice as a result of defective retrograde IFT.41 ,42 The IFT dynein-2 protein complex is not yet fully defined28 ,43 but besides homodimers of DYNC2H1 heavy chains,29 ,40 ,44 it likely contains the light intermediate chain DYNC2LI145–48 and intermediate chain DYNC2I1.49 Dynein light chains DYNLL1/LC850 and DYNLT1/TCTEX143 ,51 have also been associated with the IFT dynein-2 complex.
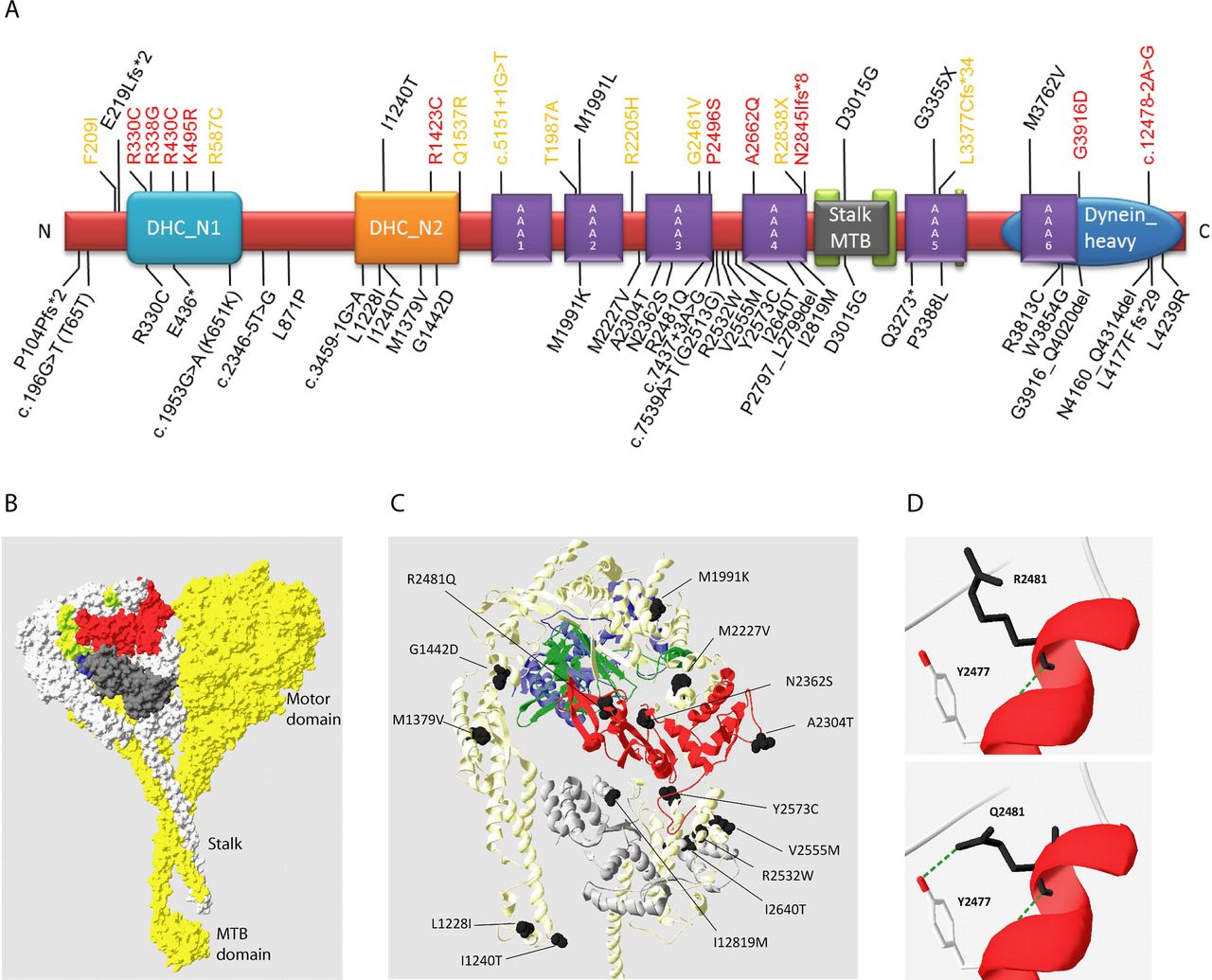
Mutations causing Jeune asphyxiating thoracic dystrophy (JATD). (A) Linear structure of the 4314 residue human DYNC2H1 protein showing the location of the 34 mutations described in this study in black, below the protein. Brackets indicate synonymous change associated with a splice site mutation. All previously reported DYNC2H1 mutations are shown above the protein, associated with JATD13 (black), short-rib polydactyly syndrome (SRPS II)24 ,25 (red) and SRPS III13 ,26 (orange). Conserved protein domains were taken from the consensus CDS entry for NP_001073932.1. The DYNC2H1 protein domains contain the six AAA+ domains of the hexomeric ring-like ATP-hydrolysing motor domain, AAA1-AAA6: AAA1 (amino acids 1651–1875), AAA2 (aa. 1938–2161), AAA3 (aa. 2251–2505), AAA4 (aa. 2617–2863), AAA5 (aa. 3244–3479) and AAA6 (aa. 3697–3912). In addition, other domain-associated structures allow DYNC2H1 to function as a motor: a thin microtubule binding stalk domain between AAA4 and AAA5 for attachment to microtubules (MT-binding stalk, aa. 2881–3227); an N-terminal tail (DHC_N1, aa. 234–676); and a linker domain (DHC_N2, aa. 1120–1520) thought to change position in different nucleotide states to create the powerstroke for motility along microtubules; plus a conserved C-terminal domain arranged on top of the ATPase ring (Dynein_heavy, aa. 3621–4311).30 ,31 (B) The predicted human DYNC2H1 protein is shown modelled to the resolved crystal structure of the cytoplasmic dynein heavy chain of Dictyostelium discoideum (DYHC_DICDI; PBD 3VKH)32 using Swiss-Model.33 Amino acid residues 1204–2969 could be modelled with confidence; AAA1, blue; AAA2, lime green; AAA3, red; AAA4, dark grey. The chain B of 3VKH used for the modelling is highlighted in light grey. (C) The location of the DYNC2H1 missense mutations that map to the regions of the protein that were possible to model by homology are shown in black. (D) The p.R2481Q substitution missense mutation could create a new hydrogen bond between Q2481 and the conserved tyrosine (TYR) at position Y2477.
Several factors have historically impeded genetic diagnosis in SRPS such as JATD, including extensive underlying genetic heterogeneity, small family sizes due to perinatal lethality and recently the large size of the DYNC2H1 gene (90 exons) which make diagnostic analysis by Sanger sequencing expensive. Next generation sequencing (NGS) approaches of exome sequencing and targeted ciliome resequencing have more recently started to impact this field.8 ,9 ,12 Here, we investigated the contribution of DYNC2H1 to JATD by mutational analysis in 57 families (71 affected patients) from two centres at UCL-ICH London and Radboud University Nijmegen Medical Centre, using a combination of single nucleotide polymorphism (SNP)-based homozygosity mapping, exome NGS and Sanger sequencing. We report a total of 34 mutations affecting 19/57 (33%) of families, and only two of these mutations have previously been associated with JATD. We show that DYNC2H1 is a major cause of JATD, particularly affecting North Europeans, causing a predominantly thoracic phenotype. We also show that imaging of IFT in patient-derived cells potentially provides a useful diagnostic tool. The study confirms the potential of high throughput NGS to effectively identify the molecular basis of rare genetically heterogeneous diseases such as JATD, and to provide important new genotype–phenotype data that were previously very difficult to obtain.
Methods
Patients
Inclusion criteria for the study were clinical and radiological signs of JATD (short ribs/ narrow thorax, small ilia with acetabular spurs, short long bones and brachydactyly), and additional features such as renal disease, liver disease, retinopathy and polydactyly if present. The UCL samples had also all been excluded for IFT80 mutations. A total of 71 JATD patient samples (61 UCL, 7 Nijmegen) fulfilling these criteria were processed by exome sequencing, and three Nijmegen samples were screened for DYNC2H1 mutations by Sanger sequencing. All blood and skin biopsy samples were obtained with informed consent according to the approved guidelines of the local ethics committees and review boards.
SNP array and CNV analysis
Genotyping of 262 000 SNPs was performed using 250 K NspI Gene Chip Arrays (Affymetrix) according to the manufacturer's instructions, with genotypes extracted using Genotyping Console software (Affymetrix). PLINK52 was used homozygosity mapping and Copy Number Analyzer from Affymetrix GeneChip (CNAG) V.2 software53 for copy number variation (CNV) detection. All ‘UCL’ prefix London samples were analysed for CNVs using the ExomeDepth Program.54
Whole exome sequencing
The exomes of patients JATD-1 and -2 were targeted with the Agilent SureSelect kit (Agilent V.1, 38 Mb) and sequenced on a quarter of a SOLiD four sequencing slide. Reads (50 bp fragments) were aligned to the Hg18 reference genome using Bioscope V.1.3, after which variants were called by the diBayes programme. Sample JATD-3 was targeted with the SureSelect kit (Agilent V.2, 50 Mb) and sequenced on a Life Technologies 5500XL instrument. Reads (50 bp fragments) were aligned to Hg19 reference genome, followed by variant calling using Lifescope V.2.1 software. London samples (prefixed ‘UCL’ in table 1) were either processed as part of the UK10K project for exome sequencing as previously described using the Agilent V.2 50 Mb All Exon kit56 or using the Nimblegen V.3 kit (Axeq Technologies). Variants from all samples were annotated and prioritised to identify pathogenic mutations as previously described for Nijmegen samples8 ,22 and London samples.56 Inclusion criteria for variants consisted of being covered by at least 10 sequence reads wherein the variant must be present in at least 20% of the reads (variants detected in 20%–80% of the sequence reads were considered heterozygous). Variants annotated in dbSNP132, the 1000 Genomes project, the Seattle exome database (http://www.evs.gs.washington.edu) or in our inhouse databases with an allele frequency above 0.5% were removed. An autosomal recessive inheritance model was applied for gene identification, with known JATD and SRPS genes manually analysed using the Integrative Genomics Viewer (http://www.broadinstitute.org/igv/). Candidate pathogenic variants were validated and assessed for familial segregation by Sanger sequencing.
Clinical information for DYNC2H1 JATD patients
Sanger sequencing
All 90 DYNC2H1 (NM_001080463.1) exons not covered by whole exome sequencing (WES) or where the 250K SNP array indicated DYNC2H1 as a primary candidate disease gene were analysed by Sanger sequencing. DYNC2H1 primer pairs used have previously been published.13 PCR products were purified, sequenced and analysed as previously described.56 ,57
Immunofluorescent microscopy
Patient and control fibroblasts obtained from skin biopsies under signed informed consent were seeded on cover glasses and grown for 24 h in Dulbecco's Modified Eagle Medium (DMEM) with 20% fetal calf serum (FCS), then serum starved in DMEM with 0.2% FCS for 48 h. Cells were stained with antibodies against IFT88 (rabbit polyclonal, 1:100, kindly provided by G Pazour and rabbit polyclonal 1:200, 13967-1-AP from Proteintech), IFT57 (rabbit polyclonal, 1:250, kindly provided by G Pazour), acetylated α-tubulin (mouse monoclonal, 1:1000, T6793 from Sigma Aldrich) and RPGRIP1L (guinea pig polyclonal; SNC040, 1:500) as previously described.57 Secondary antibodies used were Goat-anti-Mouse Alexa fluor 405, Goat-anti-Guinea pig 568 (both from Invitrogen, 1:500) and Goat-anti-Rabbit Alexa fluor 488 (Molecular Probes, 1:500). After staining, coverslips were mounted with a drop of Vectashield (Vector Laboratories) and imaged on a Zeiss Axio Imager Z1 fluorescence microscope (Zeiss) with ApoTome attachment. Images were processed using AxioVision (Zeiss), Adobe Photoshop CS4 and Adobe Illustrator CS4 (Adobe Systems). A minimum of 100 cells per sample was assessed for cilia length and localisation of IFT proteins.
Web resources
▸ Online Mendelian Inheritance in Man (OMIM) (http://www.ncbi.nlm.nih.gov/Omim)
▸ University of California, Santa Cruz (UCSC) Genome Browser (http://www.genome.ucsc.edu)
▸ The Consensus CDS (CCDS) project (http://www.ncbi.nlm.nih.gov/projects/CCDS/)
▸ PLINK (http://pngu.mgh.harvard.edu/~purcell/plink/)
▸ Copy Number Analyzer for Affymetrix GeneChip (http://www.genome.umin.jp/)
▸ GraphPad, Quick Calcs (http://www.graphpad.com/quickcalcs/chisquared2.cfm)
▸ Polyphen2 (http://genetics.bwh.harvard.edu/pph2/)
▸ Mutationtaster (http://www.mutationtaster.org/)
▸ Sorting intolerant from tolerant (SIFT) (http://sift.jcvi.org/)
▸ Genomic evolutionary rate profiling (GERP) (http://mendel.stanford.edu/SidowLab/downloads/gerp)
▸ PhyloP (http://genome.ucsc.edu)
▸ Alamut (http://www.interactive-biosoftware.com/software/alamut/features)
Results
DYNC2H1 mutations are a major genetic cause of JATD
In total, we detected causative (biallelic) DYNC2H1 mutations in 29/71 patients (41%) from 19/57 families (33%) with JATD. The patient's clinical information is provided in table 1, and the detected mutations are summarised in table 2. We validated all DYNC2H1 variants identified by exome sequencing with Sanger sequencing, and tested whether the variants segregated with disease in all families. In all cases the segregation pattern was consistent with a recessive inheritance pattern (see online supplementary figure S1), and none of the mutations were identified in 500 inhouse control exomes with an allele frequency >0.005. Although the majority of these families were solved directly through exome sequencing, DYNC2H1 was initially not targeted in the Agilent V.1 (38 Mb) exome kit. Still, sufficient coverage was generated for 30% of the DYNC2H1 exons due to the capture by enrichment probes targeting homologous regions (ie, coverage was generated for regions that were not contained in the exome design). Somewhat surprisingly, this resulted in the detection of a heterozygous p.D3015G variant (table 2) in JATD-1 and -2, through capturing the correct regions despite their not being targeted. This mutation was validated by Sanger sequencing using a locus specific and relatively long (450 bp) PCR amplicon.
Identified DYNC2H1 mutations
With the improved Agilent V.2 50 Mb kits, the entire DYNC2H1 gene was targeted, and this significantly improved coverage to approximately 90% of all coding exons, using a ‘calling-on-target’ strategy. A comparison of coverage across the DYNC2H1 gene for the different exome sequencing kits used for screening is shown in online supplementary figure S2. In parallel, targeted Sanger sequencing of the initially non-covered DYNC2H1 exons was performed, which revealed mutations previously missed by WES in patients JATD-1, -2 and -10 (table 2). These data show that WES, despite its revolutionary improvements for gene sequencing, also poses challenges for mutation identification in research and DNA diagnostic settings.
All Nijmegen samples were analysed using SNP microarray analysis prior to exome sequencing and this had excluded larger CNVs in all patients apart from one deletion of exons 81–83 identified in patient JATD-6, who additionally carried two heterozygous missense changes identified by Sanger sequencing. In comparison with Sanger sequencing, NGS data also offers the opportunity to identify CNVs; London exomes underwent CNV analysis using ExomeDepth54 and we identified a large heterozygous deletion encompassing the last three exons of DYNC2H1 exons in patient JATD-7, in addition to a missense mutation detected by conventional WES analysis (see online supplementary figures S2 and S3, table 2). Finally, we also identified mutations in the consanguineous family JATD-9 through SNP array-based homozygosity mapping followed by targeted Sanger sequencing.
Mutations causing nonsense, frameshift and essential splice site changes were judged to be disease causing, and the identified variants were also assessed for their pathogenic potential using five different bioinformatics tools (see online supplementary table S1). These incorporate evolutionary conservation of residues to gauge their functional importance; corresponding multispecies alignment of all variants is shown in online supplementary figure S4. While most mutations are clearly predicted to impair protein function by most of the tools used, three missense alleles gave more equivocal results: p.R2481Q, p.M2227V and p.M1379V. p.R2481Q is rather poorly predicted for pathogenicity; however, there is high evolutionary conservation at this residue (see online supplementary figure S4) and it is carried in JATD-3 in combination with a nonsense change. The effect of p.R2481Q was modelled onto the predicted DYNC2H1 protein, showing that substitution of a glutamate at this residue potentially significantly affects the tertiary structure, leading to a new hydrogen bond between Q2481 and the conserved tyrosine at position Y2477 (figure 1C). p.M2227V in JATD-17 was found as a homozygous change and p.M1379V in JATD-11 as a heterozygous change. Protein modelling of these substitutions on the tertiary model of DYNC2H1 did this not reveal any suggested mechanism for their pathogenic effect (data not shown). Both are predicted as benign by Polyphen-2 but as deleterious by SIFT and predicted to create new splice donor sites by Mutation Taster; moreover, the second allele in JATD-17 is a nonsense allele (p.L4177Ffs*29) and in JATD-11 is in an early and essential splice site predicting abolishment of the exon 1 splice donor.
The heterozygous p.P2797_L2799del in JATD-15 creates an inframe loss of three amino acids which cannot be scored by the software, but these residues proline-alanine-leucine (PAL) are highly conserved and thus their loss is likely to be functionally important (see online supplementary figure S4). Finally, variants in JATD-16 (c.7437+3A>G) and JATD-18 (c.2346−5T>G) are the only splice site mutations not affecting a 100% conserved splice site nucleotides. However, both still affect a highly conserved splice site nucleotide, are predicted by Mutation Taster to cause splice defects, and also have significant GERP, SIFT and PhyloP conservation scores; moreover, the second variant in both cases is strongly predicted to be pathogenic. Patient material was not available from either family, preventing an assessment of the relevance of these variations at the RNA level.
DYNC2H1 mutational spectrum in JATD
The DYNC2H1 mutation distribution is summarised in table 2 and figure 1. A total of 34 likely pathogenic DYNC2H1 variants were identified as likely to be causal in 29 patients from 19 families. These comprised six splice site mutations and seven nonsense/deletion/frameshift ‘null’ mutations that were never seen in combination together, in addition to 21 missense changes (figure 1A). Overall, 30/34 mutations were private, while three have been previously reported: p.D3015G and p.I1240T each in one JATD patient13 and p.R330C in an SRPS II patient.24 The p.D3015G mutation seems to occur only in North European patients so far, and could represent a founder allele in this population. p.Glu436*, not previously reported, was present in two unrelated Dutch families JATD-1 and -4.
Biallelic mutations were identified in 16/19 families, but more than two were identified in JATD-5, -6 and -17. The JATD-5 allele p.L1228I is a polymorphism not annotated as pathogenic (rs189806840), and therefore is the most likely to be excluded as a disease cause. JATD-6 carried the common mutation p.D3015G, and a large deletion p.G3891_Q4020; the third variant is a heterozygous missense change p. R3813C. Together with a well-supported heterozygous frameshift mutation p.L4177Ffs*29, JATD-17.1 carries a homozygous missense p. M2227V.
The 21 missense mutations were distributed along the length of DYNC2H1 (figure 1A); however, some clustering was evident since the majority (16/21) were localised to defined functional heavy chain protein domains, mainly the AAA+ motor domain, rather than to protein areas in between defined domains (figure 1A). This clustering focused around the AAA3 and AAA4 domains which contained 8/21 missense changes (38%), with fewer in AAA2, AAA5 and AAA6, and none present in AAA1. Fisher's exact test was used to evaluate the missense mutation distribution, comparing windows of 100 subsequent amino acids along the DYNC2H1 protein. This revealed a statistically significant enrichment (p<0.01) of mutations between the AAA3 and AAA4 domains (see online supplementary figure S5), although after correcting for multiple hypothesis testing using False Discovery Rate this enrichment is no longer statistically significant (see online supplementary figure S5). We compared the DYNC2H1 mutation distribution with that of DNAH5 which is mutated in a different ciliopathy, primary ciliary dyskinesia, where mutations are considered to be highly deleterious in their effect leading to complete loss of the DNAH5 protein.35 ,58–60 The distribution of DNAH5 missense mutations mirrored that of DYNC2H1 since they also clustered to the functional domains of DNAH5 (p<0.01), and similarly this significance was lost after False Discovery Rate multiple hypothesis testing correction (see online supplementary figure S5).
The total number of DYNC2H1 missense mutations analysed are too small to make robust conclusions; however, this trend in the findings warrants further investigations to shed new light on the functional role of DYNC2H1 in JATD. Figure 1C illustrates this tendency of DYNC2H1 missense mutations to localise within the known functional domains in a 3D model of the protein, including the AAA domains. It was possible to determine the location of 14 of the missense changes within the tertiary structure of the protein (figure 1C) by homology modelling to the most advanced crystal structure of the protein, which has been determined in Dictyostelium discoideum (figure 1B).32
Variable thoracic phenotype severity in JATD patients with DYNC2H1 mutations
DYNC2H1 mutations in our genetically defined JATD cohort primarily result in defects of the skeleton. The clinical course was dominated by abnormal bone development (figure 2A–E), universally affecting the thorax cage due to a reduced rib length. Typical handlebar clavicles were observed (see online supplementary figure S6). The scoliosis evident in JATD-1 and -2 could be part of the primary skeletal phenotype; this should thus be regularly monitored (figure 2J). Several patients have brachydactyly with cone-shaped epiphyses but this was not universal; for example, brachydactyly and cone-shaped epiphyses were absent in JATD-1 and -16 (see online supplementary figure S6). Since cone-shaped epiphyses tend to only be visible after the first year of life, the age at observation becomes a factor when estimating their incidence; both JATD-1 and -16 did not have hand x-rays as adults, so we cannot exclude their later development. Bilateral polydactyly was not observed (JATD-16 had unilateral postaxial polydactyly). Although short stature with variable shortening of the limbs accompanied by some bowing was frequently found in early childhood, in adolescence and adulthood average height was reached in most cases. Most patients had pelvic abnormalities with small ilia and typical acetabular spurs. Strikingly, we observed a high variability in rib shortening ranging from a slightly narrowed chest through to severe narrowing leading to death in infancy, with a consequently variable degree of respiratory problems (figure 2F–I and table 1). These differences were observed between patients with different DYNC2H1 mutations, and also between siblings with identical DYNC2H1 genotypes, for example, in JATD-3, -18, -19 and JATD-14 (Table 1, figure 2H,I). Some cases showed an improvement of the disproportionate thorax with age (figure 2M–Q and online supplementary figure S6).

Clinical features of DYNC2H1 patients. (A–E) Hallmarks of Jeune asphyxiating thoracic dystrophy (JATD): (A, JATD-5; B, JATD-16) Small thorax due to short ribs; (A, JATD-5, B, JATD-16, C, JATD-5, D, JATD-14) Small ilia with acetabular spurs; (C, JATD-5, D, JATD-14) Shortening of femurs, accompanied by bowing in (D, JATD-14); (E) 3D reconstruction of CT images of patient JATD-4. (F–I) Severity of the rib shortening varies between different patients from different families carrying DYNC2H1 mutations as well as between affected siblings: while patient JATD-5 presents with extremely shortened ribs (F), patient JATD-18 (UCL62.2) is only mildly affected (G). (H, I) Patient JATD-14 (H, UCL80.1) is markably more severely affected than his sister JATD-14 (I, UCL80.2). (J–L) Additional features: (J) scoliosis in JATD-2, (K) syndactyly in JATD-2, (L) ear malformation in JATD-16. (M–Q) Thoracic narrowing becomes less pronounced with increasing patient age. (M) Shows patient JATD-16 at under 5 years; the same patient is shown a few years later in (N) at under 10 years. (O) Patient JATD-3 in his 20s, (P) patient JATD-2 in his late teens, (Q) patient JATD-1 in his mid-20s these cases have less pronounced thoracic phenotypes compared to birth or infancy, as described in the text. Note also that shortening of the upper limbs seems less severe when JATD patients reach adolescence.
Low incidence of renal or retinal disease in JATD patients with DYNC2H1 mutations
Although there are several older patients (young adults or adolescents) in our cohort, we identified renal and/or retinal abnormalities in only two affected individuals from families JATD-3 and -5. These were both generally more severe cases in whom these features were detected at a very young age, in the second month of life (JATD-3) and in utero (JATD-5). The finding that neither kidney disease nor retinopathy occur frequently in JATD patients with DYNC2H1 mutations in our cohort seems to contrast with clinical findings in JATD patients carrying mutations in IFT140, where a high frequency of kidney disease and retinopathy has been found.9 ,12 However, as some of our DYNC2H1 patients represent terminated fetuses and not all patients have reached adulthood, we cannot exclude the possibility that additional symptoms may develop in the future. Mild liver disease was diagnosed in JATD-5 and -16, and bile duct proliferation and portal tract fibrosis in JATD-3 (table 2). This overall lack of significant renal, hepatic and pancreatic involvement is consistent with previously reported JATD DYNC2H1 cases.13 There were other features in the cohort but these have not been associated with JATD and are most likely unrelated, including cardiac septum defects, oesophageal atresia, albinism, unilateral ear malformation, bilateral toe syndactyly, patent ductus arteriosus and testicular hydrocele (figure 2K,L).
IFT defects in ciliated fibroblasts from patients with DYNC2H1 mutations
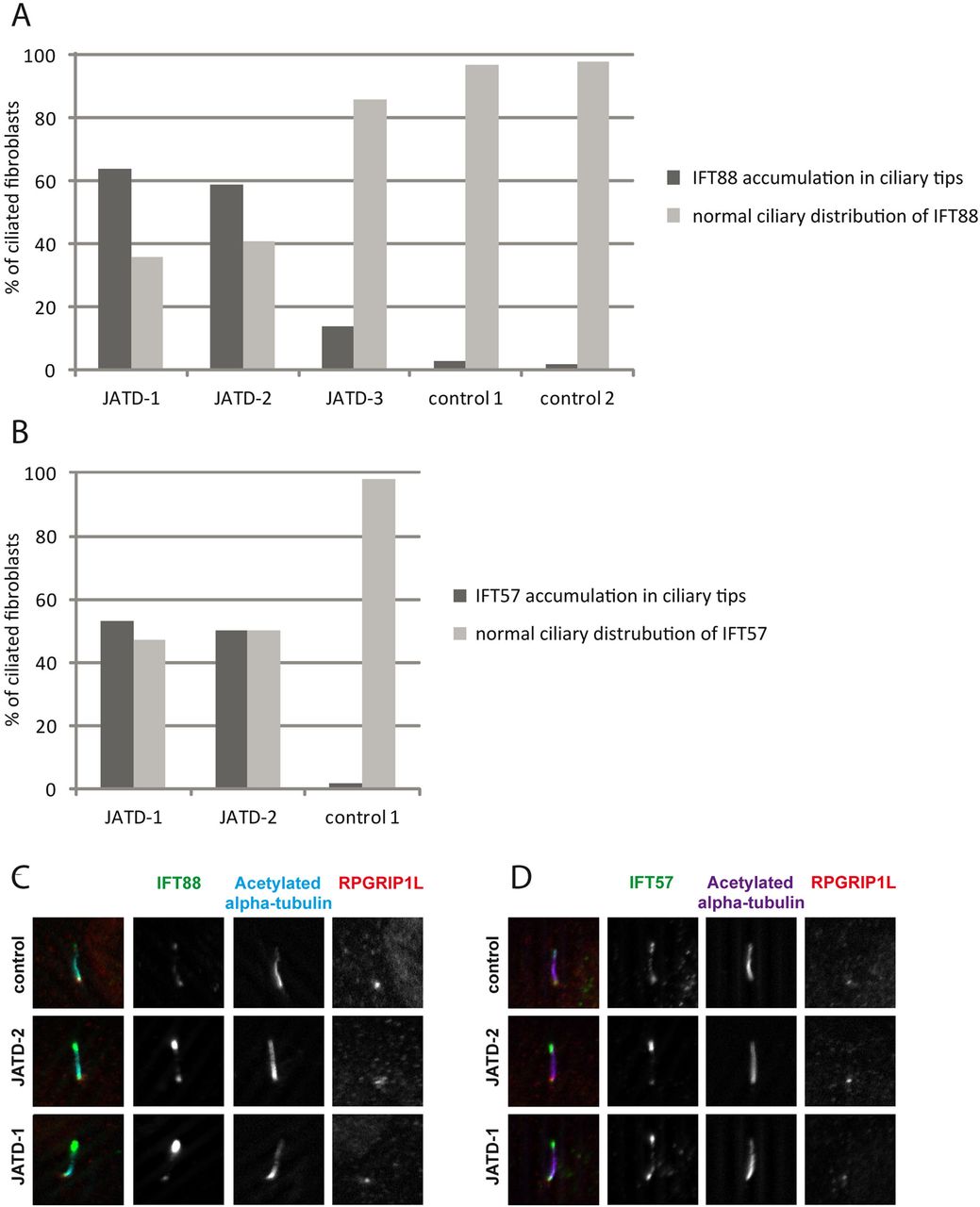
Accumulations of anterograde transport proteins in ciliary tips are typical cellular defects observed when retrograde IFT transport is disrupted. We previously showed accumulations of the IFT-B proteins IFT57 and IFT88 in fibroblasts derived from patients with skeletal ciliopathies due to mutations in IFT43, WDR35/IFT121 and WDR19/IFT144 that are all part of the IFT-A protein complex.8 ,21 ,22 We also found this cellular phenotype in three of the JATD patients with defined DYNC2H1 mutations in this study, consistent with a disruption to IFT-A retrograde transport due to DYNC2H1 deficiency. However, we saw different severity across samples. Whereas accumulations of IFT-B proteins IFT57 and IFT88 occurred in 50%–60% of fibroblasts derived from patients JATD-1 and -2, only 15% of cells were affected in JATD-3 (figure 3A–D, online supplementary figures S7 and S8). There was no major effect on ciliary length compared with controls (patients JATD-1, -2, -3 and -16, data not shown).
{kind=link}
{kind=link}
{kind=link}
Intraflagellar transport (IFT-B) accumulations in ciliary tips in Jeune asphyxiating thoracic dystrophy (JATD) patient fibroblasts. (A) In contrast to wild type fibroblasts (controls 1 and 2) in which IFT88 localises primarily to the ciliary base (and to a much lesser amount to the tip), IFT88 concentrates distally in cilia in fibroblasts derived from JATD-1, -2 and -3 family patients. Cells that were analysed in JATD-3 are derived from affected individual II-4 (Table 1). Per condition at least 100 cells stained for IFT88, acetylated-α tubulin (marker for the ciliary axoneme) and RPGRIP1L (marker for the ciliary base) as displayed in panel C were independently analysed by two blinded researchers. The control fibroblast lines were derived from individuals unrelated to our JATD patients. The graph shows that 64%, 59% and 14% of cells from patients from families 1, -2 and -3 demonstrate IFT88 concentrations in ciliary tips, whereas this effect is only observed in 3% and 2% of the cells from controls 1 and 2, respectively. (B) Another IFT-B complex partner IFT57 also accumulates in ciliary tips of JATD fibroblasts. While 53% and 50% of ciliated fibroblasts from JATD-1 and -2 patients demonstrate IFT57 accumulations in the ciliary tips, only 2% of the cells of the control display this cellular phenotype (also see panel D). Cells were analysed as per (A). (C) Compared with controls, IFT88 accumulates in distal ends of cilia in fibroblasts from JATD-1 and -2 patients. The images show a single cilium per patient or control in detail. Cells were stained with anti-IFT88 (green); antiacetylated α tubulin (marker for the ciliary axoneme, cyan); and anti-RPGRIP1L (marker for the ciliary base, red). Whole-field images displaying multiple cilia are available in online supplementary figure S7. (D) Like IFT88, IFT57 collects distally in cilia in fibroblasts from JATD-1 and -2 patients. Cells were stained with anti-IFT57 (green); antiacetylated α tubulin (purple); and anti-RPGRIP1L (red). Whole-field images displaying multiple cilia are available in online supplementary figure S8.
Discussion
To our knowledge, this is the largest WES-related study performed in JATD so far, comprising 71 patients from 57 families. Our data indicate that DYNC2H1 is the most frequent overall cause of JATD, accounting for 33% of the families screened in this study. The high mutation frequency in JATD may be influenced by the size of the DYNC2H1 protein and its central role in ciliary IFT. Since none of the DYNC2H1 patients have many extra-skeletal findings, the frequency could also reflect a higher survival rate of DYNC2H1 patients compared with those carrying mutations in other JATD-related genes.
We identified 34 DYNC2H1 mutations, 30 of which were novel, and found these to occur more frequently in JATD patients of Caucasian and Turkish origin than in patients of African and Asian ethnic background. While DYNC2H1 mutations account for 54% of the Caucasian cases we screened, and 38% of screened Turkish cases, we only detected one case of biallelic DYNC2H1 mutations in other ethnicities (15 cases screened, representing <10% of total cases, data not shown). This might partly be due to European founder effects, at least for p.D3015G, which we found in three Dutch families and which was previously reported in a French family,13 and perhaps also for two other mutations, that is, p.I1240T, which was detected in German and French families that are described in this study and previously,13 and p.E436*, which we identified in two Dutch families here. One other mutation was also found repetitively, p.R330C, albeit in families of different geographical origin: a Turkish JATD family in this study and previously in a SRPS type II patient of Haiti origin.24
Considering only the DYNC2H1 missense mutations, which can indicate important functional clues about disease causing mechanisms, the majority that we and others have described tend to localise to the known, conserved functional domains of DYNC2H1. This is similar to the mutation distribution determined in DNAH5, the heavy chain dynein gene mutated in patients with primary ciliary dyskinesia, adding support to the evidence for the DYNC2H1 variants being causal JATD alleles. Remarkably, a large number of the DYNC2H1 missense mutations localised to the region of the protein spanning AAA3-AAA4; furthermore, p.D3015G, the most common overall mutation is localised in close proximity within the microtubule-binding stalk region. In contrast, we did not identify any mutations in the AAA1 domain, which is the main ATP hydrolytic site of the DYNC2H1 motor domain and the primary driver of HC dynein's microtubular movement.61 Although AAA3 and AAA4 are very important for ATP binding, their influence on ATP binding and microtubule sliding activities is less than AAA1, and they probably play a more regulatory role.31 ,61 This importance could explain why currently no AAA1 domain mutations were identified, as variants affecting AAA1 function may be embryonic lethal in effect. A number of missense variants also cluster in the N-terminal linker region DHC-N2, which is thought to be the motile element of the dynein heavy chain, essential for its movement along the microtubules.62 ,63
These data collectively suggest that JATD DYNC2H1 missense mutations would alter protein function, but may be ‘mild’ or hypomorphic in their effect. In agreement, although seven putative ‘null’ nonsense, frameshift or deletion mutations were identified, no patients carried these in biallelic form, and none were biallelic for any of the six splice site mutations either. This presumed lack of a completely abolished protein function in surviving patients has been previously reported for lethal ciliopathy disorders including JATD.7 ,11 ,12 Experimental data using knockout mouse models indeed suggest that JATD null alleles are incompatible with mammalian development beyond mid-gestation.42 ,64
Using the IFT-B proteins IFT57 and IFT88 as markers in patient fibroblasts, we showed that retrograde IFT transport is disrupted as a result of loss of DYNC2H1 function, since these anterograde transport proteins accumulate in the ciliary tips. However, this effect was variable between JATD patient samples, consistent with a previous report.8 Thus, in the diagnostic setting it is possible that immunofluorescence studies may be of help to narrow down which aspect of IFT is deficient before the molecular defect has been defined, and second, they could provide additional evidence that a candidate gene from exome sequencing is likely the disease causing gene. However, further studies are needed to evaluate the specificity of this test. Currently we can only speculate based on the known biological roles of these proteins that we would not also find an accumulation of IFT-B particles at ciliary tips in fibroblasts of patients with such defects. Furthermore, we have not performed any comparative immunofluorescence studies in fibroblasts derived from patients with mutations in genes encoding anterograde IFT-B proteins such as IFT80, or proteins not known to be directly involved in IFT such as NEK1, to determine any specific differences in their IFT protein accumulations.
No major differences were observed in cilia number and length in fibroblasts from JATD patients, while fibroblasts from more severely affected patients with SRPS type III were previously reported to be significantly shorter and displaying bulged ciliary tips matching with defective IFT.26 Similar observations of short, bulged cilia and conclusions that this results from deficient retrograde IFT have been reported in Dync2h1 knockout mice.41 The different findings in mice compared with the DYNC2H1 patients reported here may reflect a difference between the mouse null allele and the putative hypomorphic alleles revealed in the JATD patients. In line with this is the finding that a hypomorphic Ift80 genetrap mouse which retains partial protein expression has also been found to form cilia of normal length.64 The reasons for the shorter cilia observed in more severe SRPS cases caused by DYNC2H1 mutations are a matter of speculation at this point, but could reflect a different mutation mechanism for specific DNA variants that are more deleterious to protein function, and/or may arise from an increased mutational load, that is, additional mutations in other ciliary genes influencing the phenotype in these patients.
A lack of extra-skeletal involvement was apparent for the 19 JATD families carrying DYNC2H1 mutations, apart from a few cases with renal anomalies and/or mildly elevated liver enzymes, or features that have not previously been described to be associated with JATD. Most of these features such as digit 2/3 syndactyly or unilateral ear malformation are likely to be unrelated as they occur quite frequently in the normal population, and being ‘minor birth defects’ they might often not even been reported; indeed, the 2/3 syndactyly in family JATD-2 was present in family members unaffected by JATD. Conversely, it is difficult to reconcile how these features which include a rather high frequency of mild cardiac septal defects occur as unrelated features, since we have no explanation for their occurrence. The lack of extra-skeletal involvement is consistent with the 13 DYNC2H1 patients previously reported, since renal, hepatic and pancreatic abnormalities were seen in 5/13, but only cases with severe SRPS disease, none with JATD.13 ,24–26 We also found that patients can ‘grow out’ of their thoracic and short stature phenotype as they get older, and that very often death due to respiratory failure seems to be limited to the first year of life, which is consistent with previous reports.55 JATD patients carrying IFT80 mutations also seem to exhibit a primarily thoracic phenotype,10 but apart from this observation no major phenotype–genotype correlation has yet been published for JATD. The nearly complete lack of retinal and renal disease in our DYNC2H1 JATD patients is in striking contrast to an overall high prevalence of childhood end stage renal disease and retinopathy that we and others have found in JATD and other patients who carry biallelic IFT140 mutations.9 ,12
Although many ciliary and especially IFT genes are widely expressed throughout the mammalian organism, and primary cilia are present on nearly every cell throughout the human body, ciliopathy phenotypes often seem to be restricted to certain tissues.6 ,64 ,65 The reason for this remains unclear, but it can be speculated that cell signalling pathways affected by mutations in certain ciliary genes do not have an essential function in every tissue; for example, hedgehog signalling may only cause a visible phenotype in tissues most sensitive for pathway disturbance, possibly in a time-dependent manner, such as during skeletal development. Another hypothesis could be that functionally redundant proteins can compensate for each other's loss in certain tissues.66
Our findings have important implications for clinical diagnostics, placing DYNC2H1 in a separate category from other genes encoding IFT-related proteins, in which mutations are frequently found to be associated with kidney disease and retinal degeneration.6 ,14 ,65 However, a striking variability of thoracic involvement between different DYNC2H1 patients and even between siblings with identical DYNC2H1 genotype was observed in this study. Thus, the rib length thoracic phenotype in DYNC2H1-associated JATD and therefore survival chances in infancy are difficult to predict, even with the knowledge of the underlying genotype. This presents a problem in a genetic counselling setting, for example, in cases of prenatal diagnosis for DYNC2H1 genotypes. At the moment, we can only speculate on the causes of this variability which could be due to other genetic factors such as modifier genes/mutational load,11 ,42 ,67 or the involvement of epigenetic, environmental and/or stochastic effects. In a similar vein, the p.R330C mutation seems to cause SRPS type II24 as well as JATD without polydactyly, suggesting the polydactyly phenotype does not solely depend on this mutation but might be influenced by modifier genes or epigenetic/non-genetic factors.
Most of the mutations we identified are private and they occur across the entire DYNC2H1 gene. There is even a striking lack of common alleles despite 5/19 families being consanguineous, three being of common Turkish origin. This implies that all 90 exons of DYNC2H1 would need to be screened in a clinical genetics diagnosis context. Depending on the screening method and the patient's ethnic origin, a preliminary exclusion of the ‘European’ mutations p.D3015G, p.R330C, p.I1240T and p.E436* and a focus on the exons that encode the identified functional protein domains would likely be desirable.
In summary, this large WES screen reveals DYNC2H1 mutations as a frequent cause of JATD, affecting about a third of all families. The mutation spectrum is enriched to DYNC2H1 functional domains, with a few recurrent alleles, and this information can help to target future genetic screening. The DYNC2H1-related phenotype does not appear to arise in the context of biallelic null-effect mutations, and is predominantly thoracic without extra-skeletal features. Immunostaining for cilia and IFT components in patient fibroblasts presents a potentially useful readout for making a ‘skeletal ciliopathy’ diagnosis and can provide a tool to functionally evaluate the significance of mutations in retrograde IFT genes. Together, these findings can help to guide clinical diagnostic and genetic counselling decisions in JATD.
Acknowledgments
We thank the patients and their families for their participation in the study, as well as the participating physicians. We thank all the participants of the UK10K RARE group, as listed in the online supplementary data file, that is part of the UK10K Consortium (uk10k.org.uk) in particular Matthew Hurles and Saeed Al Turki. We thank N Canham, N Crama, C Marcelis and Professor B Hamel for their clinical input into this project, J de Ligt for bioinformatics assistance and S van der Velde-Visser and M Kipping-Geertsema for excellent technical assistance. The IFT57 and IFT88 antibodies were a generous gift from G Pazour.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
MS, HHA, NVAMK, RR and HMM contributed equally.
-
Contributors MS, HHA, NVAMK, RR and HMM designed the study and wrote the manuscript. HHA, NVAMK, RR, MS, PJS, PLB and HMM supervised experiments. HHA, EMHFB, ZY, MMO, DA, LD, AH, CG, JAV, NR and E-JK performed whole exome analysis, mutational analyses and other experimental data. RDE performed protein modelling, VP performed copy number variant analysis and both contributed to writing the manuscript. JS and UK10K generated the whole exome sequencing data for UCL patients. AH and CG generated whole sequencing data for Nijmegen patients. Patient clinical ascertainment, clinical data and images were provided by AS-F, AK-K, NEV, MCvM, LG-N, KD, SS, DW, J-BLY, EF, EL, RCMH and HK.
-
Funding This research was funded by an Action Medical Research UK clinical training fellowship awarded to MS (RTF1411); the Dutch Kidney Foundation (IP11.58 and KJPB09.009) to HHA; the European Community's Seventh Framework Programme FP7/2009, under grant agreement no. 241955 SYSCILIA, to PLB and RR; a grant from the Netherlands Organisation for Scientific Research (NWO Vidi-91786396) to RR; Newlife Foundation to HMM (10-11/15). DA is funded by Action Medical Research and The Henry Smith Charity (SP4534). Funding for UK10K was provided by the Wellcome Trust under award WT091310.
Competing interests None.
Data sharing statement The authors will share data sets on request.
Patient consent Obtained.
Ethics approval Ethical review boards of the University of Nijmegen and the Institute of Child Health/Great Ormond Street NHS Hospital Trust.
Provenance and peer review Not commissioned; externally peer reviewed.
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Linked Articles
- Genotype-phenotype correlations