Article Text

Abstract
Background 22q11.2 deletion syndrome (22q11.2DS) is the most common microdeletion disorder, affecting an estimated 1 : 2000–4000 live births. Patients with 22q11.2DS have a broad spectrum of phenotypic abnormalities which generally includes congenital cardiac abnormalities, palatal anomalies, and immunodeficiency. Additional findings, such as skeletal anomalies and autoimmune disorders, can confer significant morbidity in a subset of patients. 22q11.2DS is a contiguous gene DS and over 40 genes are deleted in patients; thus deletion of several genes within this region contributes to the clinical features. Mutations outside or on the remaining 22q11.2 allele are also known to modify the phenotype.
Methods We utilised whole exome, targeted exome and/or Sanger sequencing to examine the genome of 17 patients with 22q11.2 deletions and phenotypic features found in <10% of affected individuals.
Results and conclusions In four unrelated patients, we identified three novel mutations in SNAP29, the gene implicated in the autosomal recessive condition cerebral dysgenesis, neuropathy, ichthyosis and keratoderma (CEDNIK). SNAP29 maps to 22q11.2 and encodes a soluble SNARE protein that is predicted to mediate vesicle fusion at the endoplasmic reticulum or Golgi membranes. This work confirms that the phenotypic variability observed in a subset of patients with 22q11.2DS is due to mutations on the non-deleted chromosome, which leads to unmasking of autosomal recessive conditions such as CEDNIK, Kousseff, and a potentially autosomal recessive form of Opitz G/BBB syndrome. Furthermore, our work implicates SNAP29 as a major modifier of variable expressivity in 22q11.2 DS patients.
- 22q11.2DS
- SNAP29
- CEDNIK
- Kousseff
- Exome Sequencing
Statistics from Altmetric.com
Introduction
Although clinically under-recognised, the 22q11.2 deletion syndrome (22q11.2DS) is the most common microdeletion disorder with an estimated prevalence of 1 in 2000–4000 live births. Individuals with 22q11.2DS most often have a classically associated 3 MB deletion. However, smaller atypical nested deletions have been reported, all of which result in a broad spectrum of phenotypic abnormalities.1 ,2 The occurrences of deletions in this region are related to the architecture of chromosome 22q11.2 and are associated with a non-allelic homologous recombination between chromosome specific low copy repeats (LCRs) or segmental duplications.3 The larger 3 MB deletion is associated with recombination between LCRs-A and D. Furthermore, although the smaller atypical nested deletions are predominantly mediated by LCRs A-B, they also include deletions involving LCRs B-D or C-D. In general, the associated clinical findings include: congenital cardiac abnormalities, palatal anomalies, and immunodeficiency in about three-quarters of patients; hypoparathyroidism in approximately half; and gastrointestinal and renal anomalies in about one-third.4–6 Additional findings can confer significant morbidity, such as autoimmune disease and skeletal anomalies, but are generally identified in only a subset of patients.7 Lastly, developmental delay, intellectual deficits, and psychiatric disorders such as schizophrenia are important features of this diagnosis.8
Although as many as 40 genes are deleted in patients with 22q11.2DS, mouse models of 22q11.2DS support a strong role for haploinsufficiency of TBX1, a T-Box gene in the A-B deleted region. TBX1 has been implicated in association with several clinical findings, in particular congenital heart disease.9 In patients with atypical deletions that do not include TBX1, the adaptor protein CRKL has emerged as a strong candidate for additional associated features.10 In addition, a significant number of patients with 22q11.2DS have less common findings such as polymicrogyria, myelomeningocele, cleft lip, and genitourinary abnormalities that cannot be explained solely by haploinsufficiency for TBX1 and/or CRKL. However, 22q11.2DS is considered a contiguous gene deletion syndrome as it has been proposed that loss of several or all genes within the region may contribute to the broad phenotype observed in patients.11
Next-generation sequencing advances, such as whole exome sequencing, enables identification of rare variants that may be damaging and therefore disease producing.12 This new technology permits examination of the genome including the non-deleted allele for mutations that may contribute to variable phenotypic expression in deletion syndromes. Thus, we utilised whole or targeted exome sequencing in patients with a 22q11.2 deletion and phenotypic features found in <10% of affected individuals and identified damaging mutations in SNAP29, the gene implicated in the autosomal recessive condition cerebral dysgenesis, neuropathy, ichthyosis and keratoderma (CEDNIK).13 ,14 SNAP29 is located within the C-D region on chromosome 22q11.2. Heterozygous mutations of SNAP29 have also been reported in association with cryptorchidism and hypospadias.15 Furthermore, single nucleotide polymorphisms (SNPs) in the promoter of SNAP29 have been associated with schizophrenia.16
SNAP29 (synaptosomal associated protein 29KDa) is a soluble SNARE protein that is predicted to mediate vesicle fusion at the endoplasmic reticulum or Golgi membranes.17 SNAP29 was shown to be highly expressed in myelinating glia18 and is required for lamellar body formation in the skin. It is also indirectly required for β1 integrin endocytosis and cell migration.19 We report that hemizygous deletions of 22q11.2, combined with damaging mutations in SNAP29, contribute to atypical clinical findings in patients with 22q11.2DS. Specifically, this combination unmasks one previously described autosomal recessive condition (CEDNIK), and may unmask and confirm another condition previously debated in the literature (Kousseff syndrome). It also may provide an explanation for overlapping features with a third heterogeneous condition (Opitz G/BBB syndrome).
Patients and methods
The study was approved by the Institutional Review Board of the Children's Hospital of Philadelphia (07-005352) with appropriate informed consent obtained on all subjects. In addition, the study data were handled in compliance with HIPAA (Health Insurance Portability and Accountability Act) regulations. We recruited individuals from The ‘22q and You’ Center at The Children's Hospital of Philadelphia, a large comprehensive multidisciplinary programme for patients with 22q11.2DS, for inclusion in a study of atypical clinical findings (present in <10% of overall cohort) at McGill University in Montreal, Canada and the University of Leuven in Leuven, Belgium. These findings initially included laryngo-tracheal-oesophageal abnormalities and limb differences, but were later expanded to include polymicrogyria, myelomeningocele, cleft lip, and genitourinary abnormalities. In total, 17 individuals with 22q11.2DS were studied (table 1). Whole exome sequencing was performed on four patients (patients 1, 5–7); targeted exome sequencing was performed on patient 4 and Sanger sequencing was used to screen for the presence of mutations within the gene SNAP29 in 12 patients. A detailed synopsis of the clinical findings in the patients with mutations in SNAP29 is provided in the supplementary data (patients 1–4).
A brief description of patients in this study
Whole exome sequencing and variant analysis
Whole exome capture was performed using the TruSeq Exome Enrichment Kit (Illumina, San Diego, California, USA), which targets 62 Mb of exonic sequences including 5′ untranslated region (UTR), 3′UTR, microRNA and other non-coding RNAs. The targeted exons were sequenced using paired-end technology (Illumina Hiseq sequencer) with read lengths of 100 bp. The generated exome sequencing data were analysed using our optimised bioinformatics pipeline as previously described.20 Briefly, the high quality trimmed paired-end sequences were aligned to the human reference genome (hg19) using Burrows-Wheeler Aligner (BWA) (v.0.5.9)21 Unmapped reads, reads mapping to multiple locations and PCR duplicates (PICARD V.1.48) were discarded in further analyses. A mean coverage of 66X (patient 1), 91X (patient 5), 78X (patient 6), and 76X (patient 7) was obtained for all consensus coding sequence exons.
The variant positions on the reference genome were determined using Samtools (v.0.1.17),22 mpileup and varFilter with the base alignment quality adjustment disabled, leading to the identification of approximately 299K (patient 1), 286K (patient 5), 265K (patient 6), and 269K (patient 7) variants. Additional filters were applied to narrow down the list of candidate rare variants. First, a minimum of two variant reads and >20% single nucleotide variants or >15% indels (small insertions or deletions) variant reads were considered for each called position. In order to remove systematic false positives and common polymorphisms, the variants were filtered against our in-house exome database (>350 exomes) and removed from further analysis if seen in more than five control exomes.
Subsequently, ANNOVAR23 was used to annotate the remaining variants according to the type of mutation, whether the variant was present in dbSNP132, minor allele frequency in the 1000 Genomes project, exome variant server (EVS), SIFT, PolyPhen-2 and PHASTCONS scores. Based on the assumption that the potential damaging variants are rare, the variants were kept in the final list if they had an allele frequency <0.05 in the 1000 Genomes database and predicted to be non-synonymous, that is, missense, nonsense, frameshift, or canonical splice site changes. A summary of the data obtained from each step of exome sequencing analysis is presented in supplementary table 1.
Targeted exome sequencing and analysis
The mutation in patient 4 was identified using a custom Agilent SureSelect XT target enrichment system to capture the ∼3 Mb interval of the intact chr22q11.2. Sequencing was performed using the Illumina HiScan SQ platform with 50 bp paired end reads. The reads were mapped and annotated using the Emory Mapper (Cutler and Zwick, unpublished data) and the results were bioinformatically filtered to help ensure a low level of false-positive nucleotide calls.
Sanger sequencing and analysis
To screen the SNAP29 gene, genomic DNA was extracted from whole blood using the Wizard Genomic DNA Purification Kit (Promega), following the manufacturer's instructions. All sequences, with the exception of exon 1, were amplified using 50 ng of genomic DNA and Platinum Taq Hifi DNA polymerase (Invitrogen), using the standard protocol and a Tm of 58°C. Exon 1 was amplified using Platinum Pfx DNA polymerase (Invitrogen), with a final concentration of 2× PCRXEnhancer Solution and a Tm of 55°C. Sanger sequencing was performed at the McGill University and Génome Québec Innovation Centre, using the forward primer on the unpurified PCR products. Resulting sequences were compared using BioEdit (http://www.mbio.ncsu.edu/bioedit/bioedit.html). Primers were designed by web-based Primer 3 (http://primer3.sourceforge.net/).
To determine the impact of novel amino acid substitutions on the SNAP29 protein, the PolyPhen-2 and MutationTaster tools were used.24 ,25
Results
To identify additional mutations that contribute to atypical clinical findings in patients with 22q11.2DS, we used whole exome sequencing to analyse the genome of four patients presenting with laryngo-tracheal-oesophageal and limb abnormalities (table 1, patients 1, 5–7). Homozygous 22q11.2 -associated variants were identified in one of the four patients sequenced. In patient 1, we identified 539 variants that passed all the filters after whole exome sequencing analysis, 14 of which were located on chromosome 22. Two variants out of the 14 were homozygous within the candidate region of 22q11.2: one frameshift insertion within the gene SNAP29, and one non-synonymous variant in the gene CLTCL1. Although a significant number of variants were identified in the remaining three patients (13 homozygous variants in the exome of patients 5; 17 in the exome of patient 6; and 45 in the exome of patient 7), none were in the 22q11.2 region (see supplementary table 1).
Patient 1 presented with a history of laryngotracheomalacia, a small patent ductus arteriosus, gastro-oesophageal reflux disease, failure to thrive and feeding difficulty requiring G-tube placement, chronic infection, polymicrogyria, and dysmorphic features including hypertelorism. In addition, he had: microcephaly, strabismus, optic nerve hypoplasia, bilateral sensorineural hearing loss, obstructive sleep apnoea, immunoglobulin G (IgG) and IgM deficiency, a unilateral inguinal hernia and undescended testis. More recently, he was noted to have palmoplantar keratoderma and ichthyosis, (figure 1: 1A–F). The homozygous frameshift insertion within SNAP29, c.388_389insGA (p.T130fs), has not previously been seen in dbSNP, 1000 Genomes Project or EVS, and was subsequently confirmed by Sanger sequencing (figure 2A–C). Sanger sequencing of parental blood DNA revealed a heterozygous insertion in the father at the same position (figure 2C), suggesting that the proband was hemizygous for the 22q11.2 chromosome, as determined by fluorescence in situ hybridisation. (FISH), and inherited a non-functional SNAP29 gene from the father and by inference a de novo deletion on the 22q11.2 chromosome inherited from his mother. Truncating mutations in SNAP29 are associated with CEDNIK syndrome, an autosomal recessive condition characterised by cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma.14 ,15 The frameshift mutation identified in SNAP29 is predicted to result in a truncated protein with 129 amino acids of the SNAP29 protein, and insertion of 17 novel amino acids before a premature stop (figure 2C).
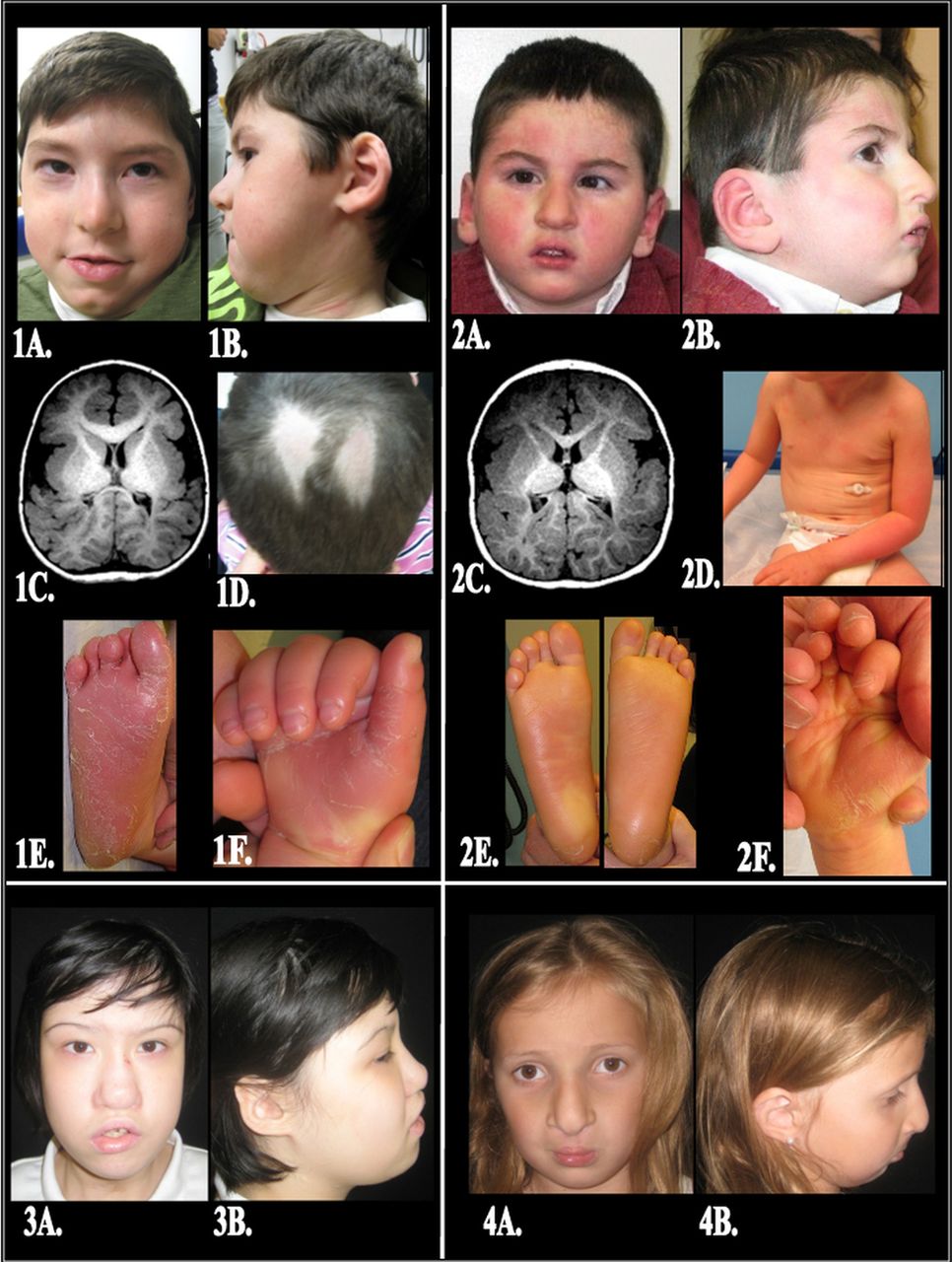
Patient description. (1A) Patient 1— Anteroposterior (AP) photo demonstrating upslanting palpebral fissures with hooded eyelids and hypertelorism; malar flatness; a bulbous nasal tip with hypoplastic alae nasi; and a mandibular cleft. (1B) Lateral photo demonstrating auricular anomalies including thick crumpled helices with attached lobes. (1C) A 2 mm thick T1 axial image of the brain reveals pronounced under-opercularisation with very wide Sylvian fissures and bilateral thick finely nodular cortex, consistent with polymicrogyria, throughout the brain but being particularly thick in the insular regions. There is abnormal brain morphology with some gyri being small and some wide. Also, there is diminished white matter and there are multiple anomalous deep fissures in both parieto-occipital regions that extend near to and deform the atria of the lateral ventricles, particularly on the right. The genu of the corpus callosum is unusually thick and the splenium is very thin. (1D) A focal area of frictional alopecia on the vertex. (1E) Right foot displays a diffuse keratoderma with erythema and thick desquamating hyperkeratotic sheets localised to the plantar surface of the foot and toes. (1F) Right hand shows diffuse keratoderma with erythema and overlying desquamation on the palmar surface, notably sparing the dorsal aspects of the fingers and nails. Accentuated skin markings can be appreciated on the volar wrist. (2A) Patient 2—AP photo demonstrating mild upslanting palpebral fissures on the left and a bulbous nasal tip with hypoplastic alae nasi. (2B) Lateral photo demonstrating normally formed ears with attached lobes; a nasal dimple; and micrognathia. (2C) A 0.9 mm thick axial T1 weighted image at the level of the insulae shows pronounced under-opercularisation with open Sylvian fissures and bilateral extensive thick nodular cortex representing polymicrogyria in all lobes of the brain, but most prominent in the insulae and the brain posterior to them. There is diminished white matter and abnormal gyral pattern throughout the brain. Also noted are anomalous deep fissures lined by the thick nodular cortex extending near to the ventricular atria and deforming them, more on the right. The genu of the corpus callosum is seen and the splenium, being very thin, is not included on this section. (2D) Anterior trunk showing diffuse xerosis and a fine, powdery, ichthyosiform scale which is accentuated on the arms. (2E) Bilateral plantar feet reveal a diffuse, glossy, yellowish keratoderma (thickening of the skin) with decreased skin markings and focal areas of desquamation on the heels. (2F) Right hand shows a diffuse, yellow-orange keratoderma with focal areas of desquamation on the palms and fingertips. (3A) Patient 3—AP photo demonstrating upslanting palpebral fissures with hypertelorism; malar flatness; a bulbous nasal tip with hypoplastic alae nasi, a blunted nasal tip with a dimple; asymmetric crying facies; and a healed tracheostomy scar. (3B) Lateral photo demonstrating auricular anomalies including crumpled helices with attached lobes and micrognathia. (4A) Patient 4—AP photo demonstrating hypertelorism; malar flatness; a bulbous nasal tip with hypoplastic alae nasi; and a repaired bilateral cleft lip and palate. (4B) Lateral photo demonstrating attached ear lobes and micrognathia.

Identification of a homozygous 2 bp frameshift insertion within the gene SNAP29 by exome and Sanger sequencing. (A) The SNAP29 gene is located on the long arm of chromosome 22 at position 22q11.2. It is 32 kb in size and is composed of five exons. (B) The grey horizontal arrows depict the 100 bp paired-end reads aligned to the positive strand of human genome (hg19) and cover the 2 bp ‘GA’ homozygous insertion at position 388_389 in exon 2. Of 19 unique reads mapped at the genomic position chr22:21224770, 17 reads displayed the 2 bp homozygous insertion. Note that the BWA (Burrows-Wheeler Aligner) placed the insertion at position c.383_384, whereas the correct HGVS (Human Genome Variation Society) notation is c.387_388dup. (C) Chromatograms show the result of Sanger sequencing in patient 1 and his parents. Patient 1 carries a homozygous 2 bp insertion resulting in a frameshift and a premature stop codon 17 amino acids downstream (p.T130fs). The father is a heterozygous carrier for the 2 bp insertion at the same position. The position of the insertion is indicated by the green arrow on the chromatograms of patient 1 and his father. In the lower part of figure, the frameshift DNA sequence and the respective translation into protein are given.
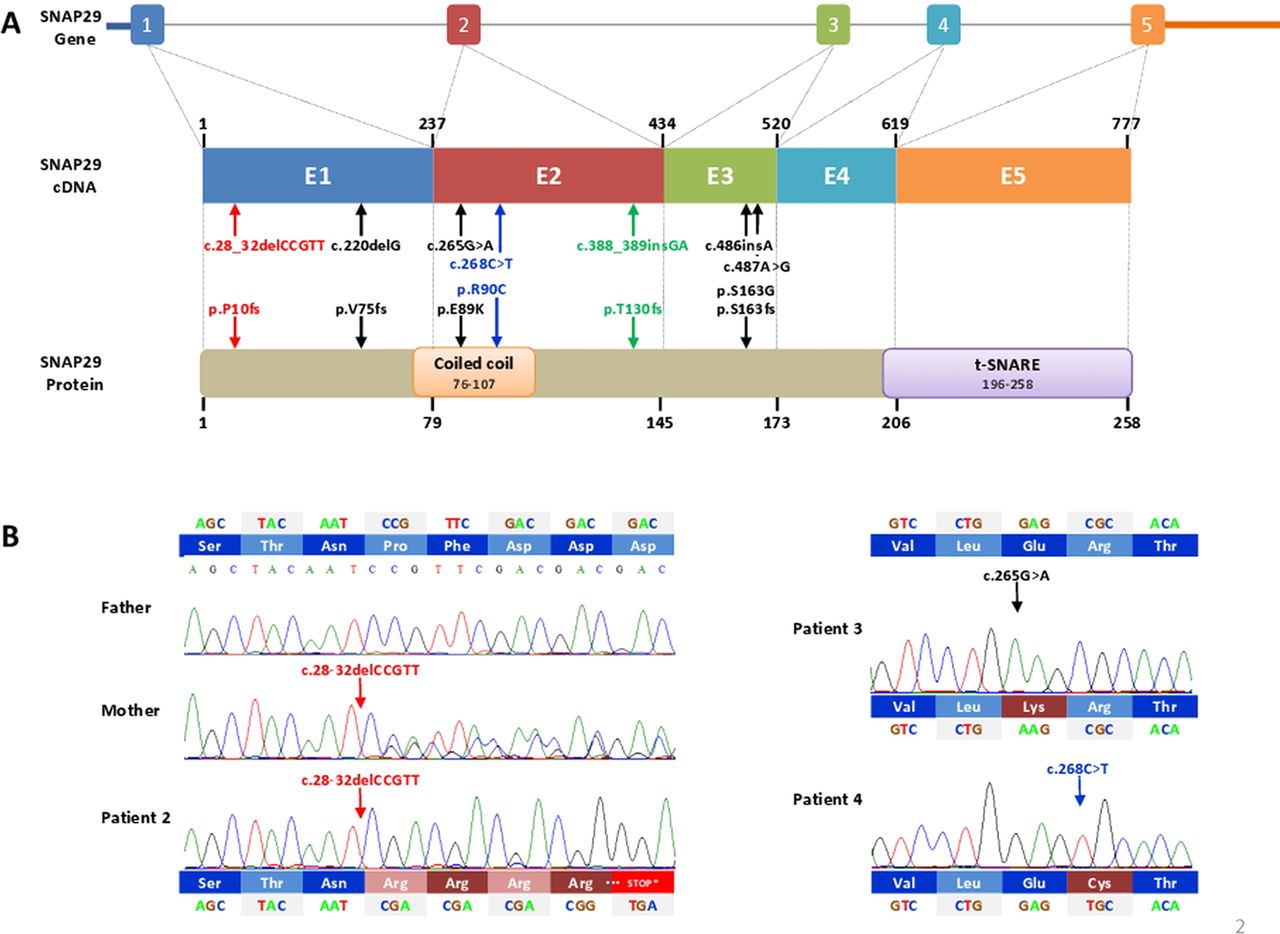
In light of this finding, we concluded that the SNAP29 mutation, in conjunction with the 22q11.2 deletion, unmasked the symptoms of CEDNIK syndrome in this patient, including diffuse polymicrogyria (similar to the 2011 Fuchs-Telem et al report14), an ichthyosiform dermatitis with secondary hypohidrosis, atopic dermatitis, and palmoplantar keratoderma. To determine if other patients with 22q11.2DS and similar atypical findings might also have mutations in this gene, we screened the coding exons 5′ and 3′ splice sites of SNAP29 in 12 additional patients by Sanger sequencing. Thereafter, we identified a 5 bp deletion in exon 1 (c.28_32delCCGTT, p.P10fs) in patient 2 (figure 3B). Atypical findings in patient 2 included features consistent with CEDNIK syndrome, as well as with patient 1: microcephaly, polymicrogyria (similar to the structural differences reported by Sprecher et al13), optic nerve hypoplasia, hypertelorism, sensorineural hearing loss, palmoplantar keratoderma, and ichthyosis; additional findings also included a preauricular tag, amblyopia, hypospadias, and type 1 diabetes (figure 1: 2A–F and supplementary data). Sanger sequencing of DNA from the parents of this patient revealed the c.28_32delCCGTT mutation in the mother's DNA (figure 3B). This deletion is predicted to result in a frameshift insertion of 42 novel amino acids before a premature stop in the SNAP29 protein (figure 3B). In addition, these studies also revealed a heterozygous A to G transition (c.317A>G) that would generate a missense (p.Q106R) mutation in the father (see supplementary figure 1). Parent of origin studies, using microsatellite markers, revealed that the proband shares the D22S264 allele with his mother, indicating the 22q11.2 deletion was paternal in origin, consistent with the SNAP29 findings.
{kind=link}
{kind=link}
{kind=link}
Schematic representation of SNAP29 gene, cDNA and protein structure. (A) Upper panel is genomic structure of the SNAP29 gene, comprises five exons (numbered 1–5) and 5′ and 3′ untranslated regions (UTRs). Introns are represented by a straight grey line. The structure of 777 bp SNAP29 cDNA is shown in the middle panel. Numbers above the cDNA diagram show the exons (named E1–E5) boundary nucleotide. The vertical dashed grey lines align the location of each exon to the regions of SNAP29 protein that each exon encodes. Numbers below the protein diagram show the contribution of each exon to the amino acid sequence. The SNAP29 protein has a length of 258 amino acid residues and is composed of two domains: coiled-coil (orange box) and t-SNARE (purple box). The position of seven mutations identified in SNAP29 is shown by arrows on cDNA and protein levels. The three mutations identified in this study are coloured in green for insertion and in red for deletion. Black arrows show the position of four previously reported SNAP29 mutations, associated with cerebral dysgenesis, neuropathy, ichthyosis and keratoderma syndrome. (B) Sanger sequencing confirmed all of the mutations identified in this study. Patient 2 and his mother are homozygous for a 5 bp deletion (c.28_32delCCGTT) resulting in a frameshift (p.P10fs) and a premature stop codon 42 amino acids downstream. Patient 3 and 4 carry a homozygous sequence variant resulting in glutamic acid to lysine (p.E89K) and arginine to cysteine (p.R90C) changes, respectively.
In a third patient (patient 3) with subglottic stenosis post-tracheostomy, cleft palate, sacral myelomeningocele, bilateral sensorineural hearing loss, tetralogy of Fallot with pulmonary atresia, hypocalcaemia, severe gastro-oesophageal reflux disease with feeding difficulty necessitating G-tube placement, asymmetric crying facies, hypocalcaemia, microcephaly, hypertelorism, clinodactyly, and hydronephrosis (figure 1: 3A,B), we identified a homozygous mutation (c.265G>A) which resulted in a missense (p.E89K) mutation in a conserved glutamic acid. This mutation was not predicted to be damaging after PolyPhen-2 analysis with a score of 0.021. However, this specific mutation was predicted to be damaging by MutationTaster (p=0.94) and has been reported to be associated with genitourinary abnormalities in a previous study by Zhang et al.15
A fourth patient (patient 4) presented with a bilateral cleft lip and palate and pseudoexotropia secondary to hypertelorism as the only atypical features. Additional findings included an atrial septal defect, mild to moderate conductive hearing loss, hypocalcaemia, and hyperextensible joints (figure 1: 4A,B). Targeted exome sequencing identified and Sanger sequencing confirmed a second point mutation in the coiled-coil domain which resulted in a missense (c.268C>T; p.R90C) mutation. This amino acid is highly conserved in all mammals but one, rodent (see supplementary figure 2), and is predicted to be damaging by MutationTaster (p=0.99) and possibly damaging by PolyPhen2, with a score of 0.890.
Discussion
Recurrent structural rearrangements in the human genome are responsible for a large number of human disorders associated with congenital abnormalities. Human deletion syndromes occur at a combined frequency of 1 in 1000 live births. Some examples include Prader-Willi,26 Phelan-McDermid,27 Williams-Bueren28 and 22q11.2DS.3 ,11 ,29 In many of these disorders, the recurrent deletion regions are defined within a certain range, the phenotypic defects constitute continuous spectra, there are a large number of genes contained within the candidate intervals, and it is unclear which gene(s) are primarily responsible for the observed phenotypic features.30 Moreover, the phenotypic spectra are often broad, and many patients present with atypical or extreme phenotypes. Previously, identifying the breakpoints of the deletions and the genes contained within the structural rearrangements constituted the limit of resolution in determining the extent of genetic involvement. This resulted in numerous unanswered questions and speculation regarding the involvement of modifier genes and epigenetic effects as possible explanations for this phenotypic variability. However, as has been recently demonstrated in the case of Van den Ende Gupta syndrome,12 ,31 large deletions may unmask the deleterious effect of mutations on the homologous allele, resulting in loss of function.
Likewise, in 1995, we described the association of Bernard-Soulier syndrome,32 an autosomal recessive condition involving thrombocytopenia and increased megakaryocytes, in a patient with 22q11.2DS, who was noted to have notably decreased ristoctein-induced platelet aggregation and GP1bB on the platelet surface by flow cytometry. Later, this was explained by a secondary mutation in the promoter region of the GP1BB gene on the remaining 22q11.2 allele that altered a GATA binding consensus site32 ,33 This mutation, in combination with the 22q11.2 deletion as is the case with the SNAP29 abnormalities observed in patients 1 and 2 and their features of CEDNIK syndrome, essentially unmasked an autosomal recessive condition. Thus, our ability to identify such events will have a great impact on understanding the causative genes underlying human deletion syndromes, as well as understanding the function of previously uncharacterised genes which contribute to the unusual and extreme phenotypes observed in a subset of patients with 22q11.2DS.
Mutations in SNAP29 are associated with CEDNIK syndrome and genitourinary malformations, and SNPs in the 5′ region have been associated with schizophrenia.16 Four different mutations were previously reported in SNAP29: a homozygous c.486insA insertion which results in production of a truncated protein (p.S163fs);14 a 1 bp deletion (c.220delG) which was predicted to also result in a truncated protein (p.V75fs) and was shown to result in loss of the protein by Western blot analysis;13 and two heterozygous mutations, c.487A>G and c.265G>A of unknown status.15 In this study, we report three novel mutations (c.388_389insGA, c.28_32delCCGTT, and c.268C>T) in SNAP29 that contribute to atypical findings in 22q11.2 patients as well as one previously reported mutation (c.265G>A) (figure 3A).
SNAP29 is a ubiquitous member of the t-SNARE proteins that forms a 4-helixes bundle with SNAP23, SNAP25, and SNAP47 during vesicle docking at the plasma membrane.34 SNAP29 has also been shown to complex with several other proteins involved in vesicle secretion such as clathrin, alpha adaptin, APAP2, EHD1, and RAB3A.34 ,35 Loss of function mutations in this gene are associated with delayed maturation and secretion of lamellar granules that are involved in the transport of lipids and proteases to the epidermis.36 More recently, SNAP29 mutant cells were also shown to have abnormal endocytosis of β1-integrin, suggesting that abnormal cell migration may be contributing to the polymicrogyria phenotype found in patients.19 Intriguingly, four of the seven mutations identified in this gene are before or in the coiled-coil domain of SNAP29 (figure 3A). This is a SNAP29 specific coiled-coil domain that is not shared by other related t-SNARE members. Since these domains are normally used for protein–protein interaction it is intriguing to postulate that mutations in this domain interfere with the efficiency of the interaction of SNAP29 with other SNARE proteins. This interference may result in abnormal interaction with specific associated SNAREs in a tissue dependent manner, thus explaining the variability in phenotypes found. In addition, although the E89K mutation is predicted to not be damaging by PolyPhen-2, this mutation is of an amino acid that is conserved in all primates and placental mammals. We postulate that this mutation is damaging as predicted by MutationTaster and thus likely to perturb SNAP29 function.
Two of the four patients described herein (patients 1 and 2) share a number of common clinical features: (1) an early-onset ichthyosiform presentation; (2) eventual development of palmoplantar keratoderma between 1–2 years of age that is accentuated at sites predisposed to pressure or friction; (3) features consistent with mild to moderate atopic dermatitis; and (4) hypohidrosis with associated heat intolerance (but without other features associated with hypohidrotic ectodermal dysplasia). The ichthyosiform skin changes and palmoplantar keratoderma are likely related to deficiency of the SNARE protein which plays a central role in Golgi function and synaptic vesicle recycling. The combination of the 22q11.2 deletion with a mutation in the remaining SNAP29 allele encoding the SNARE protein presumably leads to significantly decreased expression of SNARE. As a result, the normal release of lipids and proteases from lamellar granules into the extracellular spaces of the cornified cell layer is likely impaired, resulting in defective desquamation and barrier formation. Defective barrier function may then lead to epicutaneous sensitisation and, secondarily, features of atopic dermatitis. Although the specific mechanism behind the hypohidrosis seen in both patients and the enamel hypoplasia in one of the two is unknown, these suggest that patients with these genetic changes may share features overlapping those of ectodermal dysplasia. In addition they both have polymicrogyria, hypertelorism/relative hypertelorism, bilateral sensorineural hearing loss, and significant feeding, swallowing and airway difficulties.
Patient 3 has several significant atypical findings including subglottic stenosis, bilateral sensorineural hearing loss, cleft palate, hypertelorism, a sacral myelomeningocele, and clinodactyly, in addition to classically associated features of 22q11.2DS such as tetralogy of Fallot with pulmonary atresia, asymmetric crying facies, and hypocalcaemia. This is notable because in 1984, Kousseff37 reported the association of sacral meningocele, conotruncal cardiac anomalies, unilateral renal agenesis, and dysmorphic features in three siblings and suggested autosomal recessive inheritance. Toriello et al38 reported a similar isolated case in 1985 and coined the term Kousseff syndrome. In 2002, Forrester et al39 restudied the family reported by Kousseff and identified a 22q11.2 deletion in the proband and his father, thereby attributing the sacral myelomeningoceles to the 22q11.2DS. Thereafter, the existence of Kousseff syndrome as a distinct entity was called into question. However, in 2004, Maclean et al40 subsequently reported two patients with Kousseff syndrome, one with a 22q11.2 deletion and the other without a deletion, both of which were performed by FISH, and concluded that Kousseff syndrome is causally heterogeneous. Perhaps the features in our patient 3 also represent the unmasking of an autosomal recessive condition, Kousseff, in the same manner as CEDNIK, with a deletion of 22q11.2 on one chromosome and a mutation in SNAP29 on the other. Thus, we postulate that deletion of 22q11.2 on one chromosome associated with mutation(s) in SNAP29 may also explain the presence of myelomeningoceles, a typically uncommon associated feature in 22q11.2DS, in a subset of Kousseff37 patients.
Patient 4, presenting with a bilateral cleft lip and palate and hypertelorism also warrants further discussion as both features are reported in association with the 22q11.2 deletion, but infrequently. However it is notable that the combination of cleft lip and palate, hypertelorism, laryngotrachealesophageal anomalies and hypospadias has been described in individuals with the heterogeneous Opitz G/BBB syndrome,41 where an X-linked form – due to mutations in MID1 and an autosomal dominant form with linkage to 22q11.2, and an association with the 22q11.2 deletion syndrome have all been reported previously.42–46 These individuals typically present with laryngo-tracheal-oesophageal abnormities, as seen in three of our four patients reported here with SNAP29 mutations in combination with a 22q11.2 deletion, as well as: cleft lip and palate in one of four; hypertelorism in all four; severe swallowing difficulties/oesophageal dysmotility in three of four; and genitourinary abnormalities such as hypospadias in one of the two males and cryptorchidism in the other. Thus, SNAP29 mutations in association with a 22q11.2 deletion may also elicit a set of symptoms previously described in association with Opitz G/BBB syndrome, and perhaps unmask an autosomal recessive phenocopy. This might also explain the child with ‘apparent G syndrome’ reported by Williams and Frias,47 as well as isolated case reports such as the female described by Neri et al.48 Furthermore, although SNAP29 is known to be associated with schizophrenia we could not assess this condition in our patients. In fact, three of our four patients are below the age of onset, and the fourth patient, a young teen, is likely too impaired cognitively to assess for this phenotype.
In addition, in one patient (patient 1) we also identified a homozygous mutation in the CLTCL1 gene that is predicted to be damaging. This variant in CLTCL1 has previously been seen in the dbSNP, 1000 Genomes and three unrelated exome samples sequenced at our centre. CLTCL1 encodes clathrin heavy chain-like 1, a major component of coated vesicles.49 CLTCL1 is not found in rodents and is predicted to function species-specifically in Glut4 transport in muscle cells, although it may also have functions in other tissues. Intriguingly, although the majority of abnormalities found in patient 1 can be explained by the mutation identified in SNAP29, this patient also had an unusual B cell immunity that may more likely be the result of the mutation in CLTCL1. Sequencing of additional patients will allow us to determine if the mutation in CLTCL1 is associated with B cell abnormalities. However, since an atypical deletion that includes SNAP29, LZTR1 and P2RXL1 was recently found associated with immune deficiency, it remains possible that the mutations that we identified are responsible for this phenotype.50
In conclusion, we have found that mutations in a single gene, in unrelated patients, contribute to the phenotype of patients with a microdeletion syndrome, implicating SNAP29 as a major modifier of variable expressivity in 22q11.2DS patients. Our work confirms that the phenotypic variability observed in patients with 22q11.2DS may be due to additional mutations on the non-deleted chromosome, which in some instances unmask a previously described autosomal recessive condition such as the CEDNIK and potentially Kousseff syndromes, and in other cases simply contributes to the presence of atypical findings. In fact, the majority of our patients did not have mutations in SNAP29, suggesting that non-coding mutations in SNAP29, and mutations in additional genes in the 22q11.2 region or on other chromosomes, are contributing to atypical findings in those patients, as previously suggested by Stalmans et al.51 Future work is focused on identifying these additional mutations in the hope of stratifying patients for optimal treatment and genetic counselling.
Acknowledgments
We would like to acknowledge Patrick McMahon MD for his help in assessing and documenting the dermatologic findings in patients 1 and 2; Elizabeth Bratton for assistance with clinical data collection in the ‘22q and You’ Center; Piotr Jurgielewicz for assistance with gathering reference materials; Colleen Franconi for technical assistance in the laboratory of Dr Beverly S Emanuel, all at the Children's Hospital of Philadelphia. We also wish to acknowledge the contribution of the high-throughput sequencing platform of the McGill University and Genome Québec Innovation Centre (Montréal, Canada) and Dr Jacek Majewski for his help in analysing the exome data.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
DMM-M and SF contributed equally to this work
-
Funding This work was supported by a grant from the Canadian Institutes of Health Research (#MOP-102666) to LJM, and NIH support (HL84410, MH87636 and HD070454) to BSE, DMM-M and EHZ. BAN is supported by KOLUMB fellowship from the Foundation for Polish Science. The Charles EH Upham Chair to BSE provided funds for this research at the Children's Hospital of Philadelphia. JV was supported by the Jerome Lejeune Foundation and LJM is a member of the Research Institute of the McGill University Health Centre, which is supported in part by the FRSQ.
-
Contributors DMM-M liaised with the collaborative investigators, selected and recruited subjects for participation in the research study, oversaw sample and data collection, participated in scientific discussions surrounding the laboratory results, wrote the clinical sections of the paper including table 1, figure 1, and the respective figure legends, and drafted and revised the paper. SF performed exome sequencing data analysis, designed the primers, assembled the figures on the molecular findings, drafted and revised the paper. TR performed Sanger sequencing analysis of 14 patients, reviewed the draft and revised the paper. BAN performed chromosome 22q11.2 exome capture analysis, reviewed the draft and revised the paper. JS performed chromosome 22q11.2 exome capture analysis, reviewed the draft and revised the paper. AB arranged for sample collection and distribution, monitored data collection, obtained informed study consent, obtained parental photo consent, obtained photographs, drafted table 1 and revised the drafted paper. EM performed Sanger sequence confirmation, reviewed the draft and revised the paper. DRL participated in scientific discussions surrounding the laboratory results and clinical findings, reviewed the neurologic data on patients 1, 2 and 3, wrote the discussion on the associated neurologic phenotype, participated in scientific discussions surrounding the laboratory results, and reviewed the drafted and revised paper. AY participated in scientific discussions surrounding the laboratory results and clinical findings, reviewed the dermatologic sections of the paper, submitted dermatologic photographs on patients 1 and 2, wrote the clinical descriptions and discussion on the dermatologic section and figure 1, and reviewed the drafted and revised paper. LB reviewed the neuroimaging data on patients 1, 2 and 3, selected the brain images for figure 1, wrote the description for the MRI findings, and reviewed the drafted and revised paper. KES participated in scientific discussions surrounding the laboratory results and clinical findings, reviewed the immunologic data on all patients, wrote the postulate for the immune findings in patient 2, and reviewed the draft and revised paper. STW supervised the exome 22q11.2 capture experiments at Emory University. BSE participated in the planning and execution of the study design, participated in scientific discussions surrounding molecular findings, and reviewed the draft and revised paper. JRV supervised the exome 22q11.2 capture experiments performed by BAN, participated in discussion of the molecular findings and reviewed the draft and revised paper. EHZ initiated the collaborative project, participated in the planning and execution of the study design including subject selection, participated in scientific discussions surrounding the laboratory results and clinical findings, and reviewed the draft and revised paper. LAJ-M designed the study, liaised with the collaborative investigators, supervised the exome and Sanger sequence data collection and analysis at McGill University, participated in scientific discussions surrounding the molecular results, and drafted and revised the paper.