Article Text

Abstract
Background and aim Several studies have highlighted the association of the 12q13.3–12q14.1 region with coeliac disease, type 1 diabetes, rheumatoid arthritis and multiple sclerosis (MS); however, the causal variants underlying diseases are still unclear. The authors sought to identify the functional variant of this region associated with MS.
Methods Tag-single nucleotide polymorphism (SNP) analysis of the associated region encoding 15 genes was performed in 2876 MS patients and 2910 healthy Caucasian controls together with expression regulation analyses.
Results rs6581155, which tagged 18 variants within a region where 9 genes map, was sufficient to model the association. This SNP was in total linkage disequilibrium (LD) with other polymorphisms that associated with the expression levels of FAM119B, AVIL, TSFM, TSPAN31 and CYP27B1 genes in different expression quantitative trait loci studies. Functional annotations from Encyclopedia of DNA Elements (ENCODE) showed that six out of these rs6581155-tagged-SNPs were located in regions with regulatory potential and only one of them, rs10877013, exhibited allele-dependent (ratio A/G=9.5-fold) and orientation-dependent (forward/reverse=2.7-fold) enhancer activity as determined by luciferase reporter assays. This enhancer is located in a region where a long-range chromatin interaction among the promoters and promoter-enhancer of several genes has been described, possibly affecting their expression simultaneously.
Conclusions This study determines a functional variant which alters the enhancer activity of a regulatory element in the locus affecting the expression of several genes and explains the association of the 12q13.3–12q14.1 region with MS.
- Genome-wide
- Multiple sclerosis
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
Multiple sclerosis (MS) is a chronic autoimmune disease with a complex pathogenesis in which demyelination and neurodegeneration are the main contributors to disability.1 Susceptibility to MS is thought to be conferred by a combination of genetic and environmental factors.2 The best characterised region implicated in predisposition to MS is the Major Histocompatibility Complex on chromosome 6p21, specifically the HLA-DRB1* 15:01 class II allele, but this accounts for less than 50% of MS heritability.3 New powerful methods as gene expression profiling and genome-wide association study (GWAS) have provided evidence for new susceptibility loci implicated in MS.4–6 These comprise over 40 loci including IL2RA, CBLB, IL7R, CLEC16A, TNFRSF1A, PTGER4 and IRF8.7–9
For many risk loci, the association signals do not directly implicate a single gene and the causative role for candidate genes in the region can only be speculated. A striking number of loci have demonstrated genome-wide evidence for association in multiple, distinct autoimmune disorders.10 ,11 One of these loci is 12q13–14 which has been associated in GWAS for rheumatoid arthritis (RA), coeliac disease (CD) and MS.8 ,12–14 However, different genes have been suggested in each study based on the main associated signal. A meta-analysis of two published GWAS totalling 3393 RA cases and 12 462 healthy controls identified an association at rs1678542 localised in the KIF5A intronic region.13 Another meta-analysis of two published GWAS on CD (4533 cases and 10 750 controls) and RA (5539 cases and 17 231 controls) described the association of both diseases at rs10876993 localised in the intergenic region between B4GALNT1 and OS9 genes.14 Association at this locus was also described in a MS GWAS performed by the Australian and New Zealand Multiple Sclerosis Genetics Consortium in 1618 MS-cases. In this case, an associated single nucleotide polymorphism (SNP) (rs703842) was located at the 3' untranslated region (3' UTR) of the METTL1 gene.12 The last GWAS performed by the International Multiple Sclerosis Genetics Consortium with 10 000 MS patients also reported the association with this region at rs12368653 in theAGAP2 gene.8 In candidate gene studies, the KIF5A variant was demonstrated to be associated to MS15 and type 1 diabetes.16 Also rare variants in the CYP27B1 gene have been associated with MS.17 Other candidate-gene studies had demonstrated association of variants located at the CYP27B1 gene with type 1 diabetes18 and MS.19
The FAM119B gene has been pinpointed as the causal gene of the locus since its expression has been shown to be much lower in leukocytes of MS patients carrying the MS risk allele.20 On the other hand, since, in several studies, MS patients have lower levels of precursors of vitamin D in serum compared with controls, it has been suggested that CYP27B1, an enzyme implicated in Vitamin D activation, may be the causal gene.19
In this work we performed a fine mapping of the 12q13.3–12q14.1 region by a Tag-SNP approach to identify the polymorphism leading the association with MS. We also performed functional studies to determine the involvement of the associated MS risk variants in gene expression.
Material and methods
Study subjects and SNP genotyping
The entire data set studied consisted of 2876 MS patients diagnosed following Poser's criteria21 and 2910 age and gender matched healthy controls, mostly blood donors and staff, matched by age, gender, ethnicity and place of recruitment. Patients and control subjects were included in the study based upon written informed consent. Demographic and clinical characteristics of the sample collections have been published before15 ,22 (a summary is in the online supplementary table S1). SNPs were genotyped using the iPLEX Sequenom MassARRAY platform in the Spanish National Genotyping Center's facilities at Santiago de Compostela University (http://www.cegen.org). All DNAs ware received in the Centro Nacional de Genotipado (CEGEN) centre for genotyping in 96-well plates; the concentration of double-stranded DNA was assessed using PicoGreen (Molecular Probes). As quality control, two trios of Center for the Study of Human Polymorphisms (CEPH) samples were genotyped (CEPH Na10830, Na10831, Na12147, Na10860, Na10861, Na11984). The genotypes of these samples corresponded with the ones deposited in HapMap with no detection of Mendelian inconsistencies. Additionally, a 10% of random samples were subjected to regenotyping. The resulting data were concordant with an average accuracy over 99.9%. We performed quality filtering of both samples and SNPs to ensure robust association testing.
Ethics statement
All participants provided a written informed consent to participate in this study, which was approved by the Institutional Review Board of Hospital Regional Universitario Carlos Haya of Malaga, Hospital Clinic of Barcelona, Hospital Virgen Macarena of Sevilla, Hospital de Basurto of Bilbao, Hospital Clínico San Carlos of Madrid, Hospital Universitari Vall d´Hebron of Barcelona and Hospital Donostia of San Sebastian.
Luciferase assays
Six fragments in both orientations with potential regulatory activity, as indicated by ENCODE, containing the tagged variants, 24 inserts in total, plus a plasmid without insert, were assayed for luciferase activity. Thus, fragments I–VI were PCR amplified (the coordinates and the primer used are indicated in online supplementary table S2) using DNA from homozygous individual for each allele of rs6581155. The fragments were previously cloned in Vaccinia DNA Topoisomerase I (TOPO) T/A vector (Invitrogen) and sequenced. Then, each clone was introduced in forward and reverse orientation in pGL4.25 (luc2CP/minP) vector which contains a minimal promoter upstream of the luciferase reporter gene luc2CP. Triplicates were transfected into human Raji cells (ATCC CCL86). The Raji cell line was maintained in Roswell Park Memorial Institute (RPMI) 1640 medium, supplemented with 10% fetal calf serum, 2.0 mM glutamine, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 1 mM streptomycin, 100 U/ml penicillin, 0.05 mM 2-mercaptoethanol. Exponentially growing cells were transfected by nucleofection Amaxa Cell Line Nucleofector Kit V (Lonza) according to manufacture protocols. Briefly, Raji cells (2×106) were mixed with I–VI plasmid constructs or pGL4.25 control plasmid 2 μg of DNA and 0.2 μg of DNA of Renilla luciferase reporter gene plasmid pRL-TK(Promega) for normalising transfection efficiency. The transfected cells were cultured in 12 well-plates for 24 h. Cells were harvested and washed twice in Phosphate Buffered Saline (PBS) at 4°C. Luciferase activity was evaluated using Dual-Luciferase Assay System (Promega) according to manufacturers. For analysis, lysates were thawed on ice and the protein concentration was measured by the Bradford method. Then, 40 μg of protein from all supernatants were analysed for both firefly and Renilla luciferase activities in a luminometer F12 (Berthold Detection Systems). The following equation was used to determine the normalised fold changes in activity between tested groups:

The normalised fold changes in activity from each of the three experiments are then averaged together.
Statistical analysis
Departure from Hardy-Weinberg equilibrium for all the biallelic SNP markers was tested using an exact test.23 Raw data were analysed for comparison of allele and genotype counts using PLINK V.1.05. To avoid false-positive results due to multiple testing and considering that the SNPs analysed are not in complete disequilibrium, we applied the Benjamini-Hochberg method24 that is robust against positive dependence and controls the false discovery rate.25 In order to determine the effect and independence of variant association with MS, multiple logistic regression models were computed. Linkage disequilibrium (LD) patterns between SNPs were analysed with Haploview 4.2.26 Gender was analysed for potential differential risk contribution. We used the Breslow-Day Test to evaluate the homogeneity between gender cohorts. The age of onset with respect to the genotypes was analysed by the Kruskal-Wallis test.
Results
A tag-SNP association study of the region
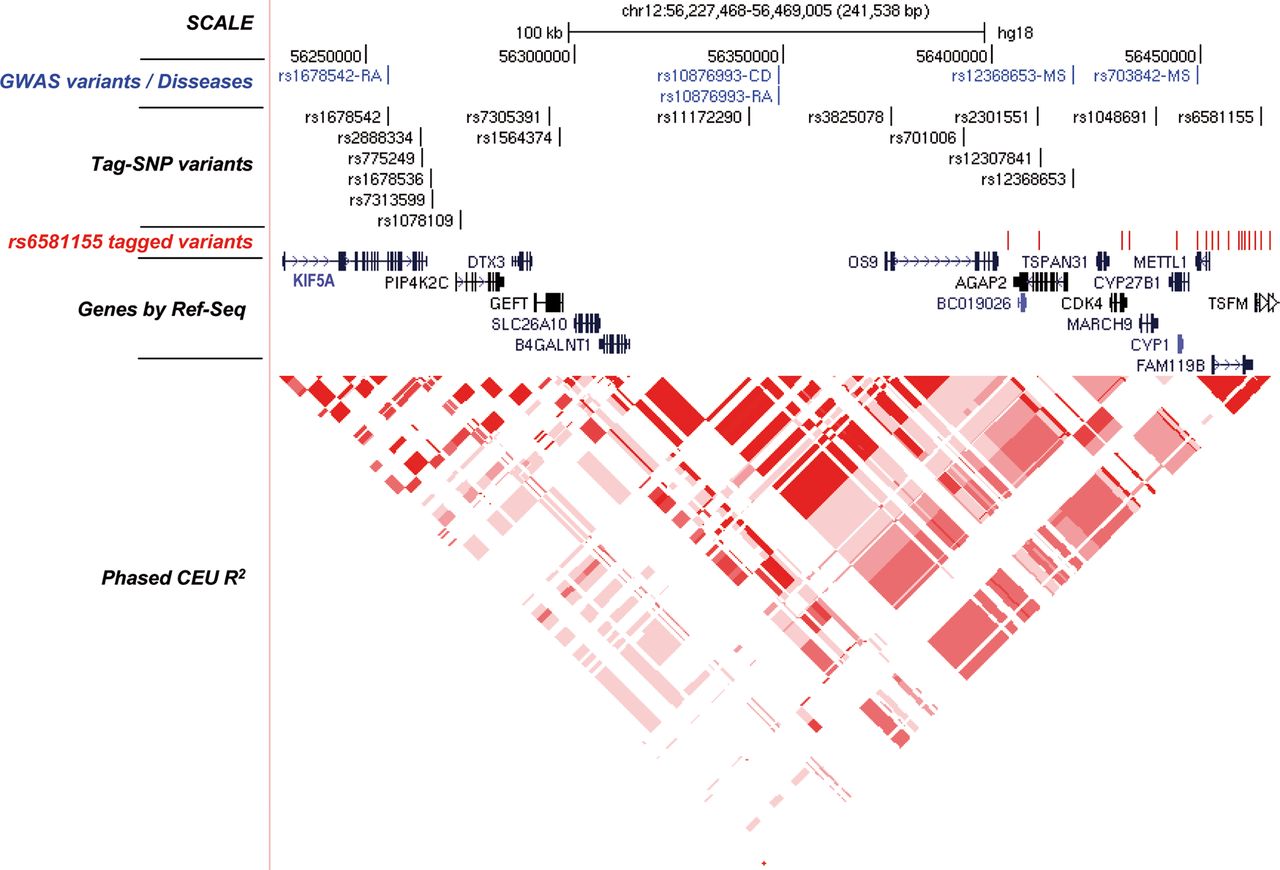
We performed the fine mapping of the 12q13.3–12q14.1 region between coordinates 56227468 to 56469005 (B36) (figure 1). To analyse this region, 16 SNPs were chosen by pairwise tagging from the HapMap B36 Utah residents with Northern and Western European ancestry from the CEPH collection (CEU) population which captured 89 markers with r2≥0.8 (mean r2=0.93) and a minor-allele frequency ≥0.1. These tag-SNPs captured the variants associated with RA, MS and CD previously reported in different GWAS (see online supplementary table S3).

Scheme of the 12q13.3–12q14.1 region. The illustration shows from top to bottom the following parts as indicated : Scale and chromosomal position (B36/gh18); (in blue) diseases-associated variants as reported in different genome-wide association study (RA, rheumatoid arthritis; CD, coeliac disease; MS, multiple sclerosis); Tag-SNPs analysed in the present work; (in red) MS-associated rs6581155-tagged variants with r2≥0.8 and a minor-allele frequency <0.1; Ref-Seq genes present in the region; the LD structure represented by r-square for the CEU population.
Tag-SNPs were genotyped in 2895 MS patients and 2942 healthy controls, mostly blood donors, matched by age, gender, ethnicity and place of recruitment. The genotypic distribution between cases and controls is shown in table 1. The genotype frequencies were fitted to an additive model. The top associated SNP was rs6581155 (p=4.57E−7; OR=0.79; 95% CI 0.73 to 0.87). We performed a logistic regression analysis to determine the effect of further independent variants, by testing the addition of each SNP to rs6581155 and by adding this polymorphism to each one of the other Tag-SNPs (table 2). The results showed that, in addition to generate the strongest signal, rs6581155 alone was sufficient to model the association in the locus.
Association study of the different Tag-SNPs with multiple sclerosis
Regression analyses of the 16 Tag SNPs
Stratification by gender and age at onset and genotypes of rs6581155 SNP showed absence of statistical significance.
Moreover, we genotyped the rare variant rs118204009 located in the CYP27B gene that has been recently associated with MS in a subgroup of 1006 patients and 1044 healthy controls.17 We did not detect this polymorphism in any individual of the MS or control groups (data not shown).
Association of rs6581155-tagged SNPs with gene transcription levels
To determine if selected Tag-SNPs could have a functional effect on the transcription levels of the genes located in the 12q13.3–12q141 region, we first used expression quantitative trait loci (eQTL) data from Zeller et al27 obtained from monocyte samples of 1490 German individuals (http://genecanvas.ecgene.net/news.php?readmore=45). There were 23 eQTLs for FAM119B, 6 for AVIL, 18 for XRCC6BP1, 18 for TSFM and 20 for TSPAN31 genes in the whole region. The variants best correlated with expression of FAM119B, AVIL, TSFM and TSPAN31were also SNPs tagged by rs6581155 and, therefore, in high LD with it (r2>0.8) (table 3). The eQTLs for the XRCC6BP gene were in low LD with rs6581155 (r2=0.18) indicating that they were not involved in MS risk. Interestingly, the minor alleles for the rs6581155-tagged SNPs, which were protective for MS, correlated with high expression of FAM119B and AVIL but low expression of TSFM and TSPAN31 (see online supplementary figure S1) although there were large differences between the correlation coefficients obtained for each gene. FAM119B was the gene most strongly correlated with the rs6581155-tagged SNPs in monocytes.
rs6581155 tagged SNPs that have been reported to be eQTLs in the region
The eQTL data from Dixon et al28 obtained in lymphoblastic cell lines (LCL) from 206 families of British descent confirmed the eQTLs previously observed for FAM119B and TSFM, although the correlation coefficients were similar for both genes in contrast to the data from Zeller et al.27 The eQTLs for FAM119B were also reported in three additional data sets carried out in different cell types, as shown in table 3.
The data from Montgomery et al,29 obtained by RNA-Seq from LCL of 60 HapMap CEU individuals, have reported a rs6581155-tagged SNP to be eQTLs for FAM119B and CYP27B1, in which the minor allele inversely correlated with high expression of these genes (correlation coefficient (r) for CYP27B1=−0.472, and for FAM119B=−0.474).
Enhancer activity
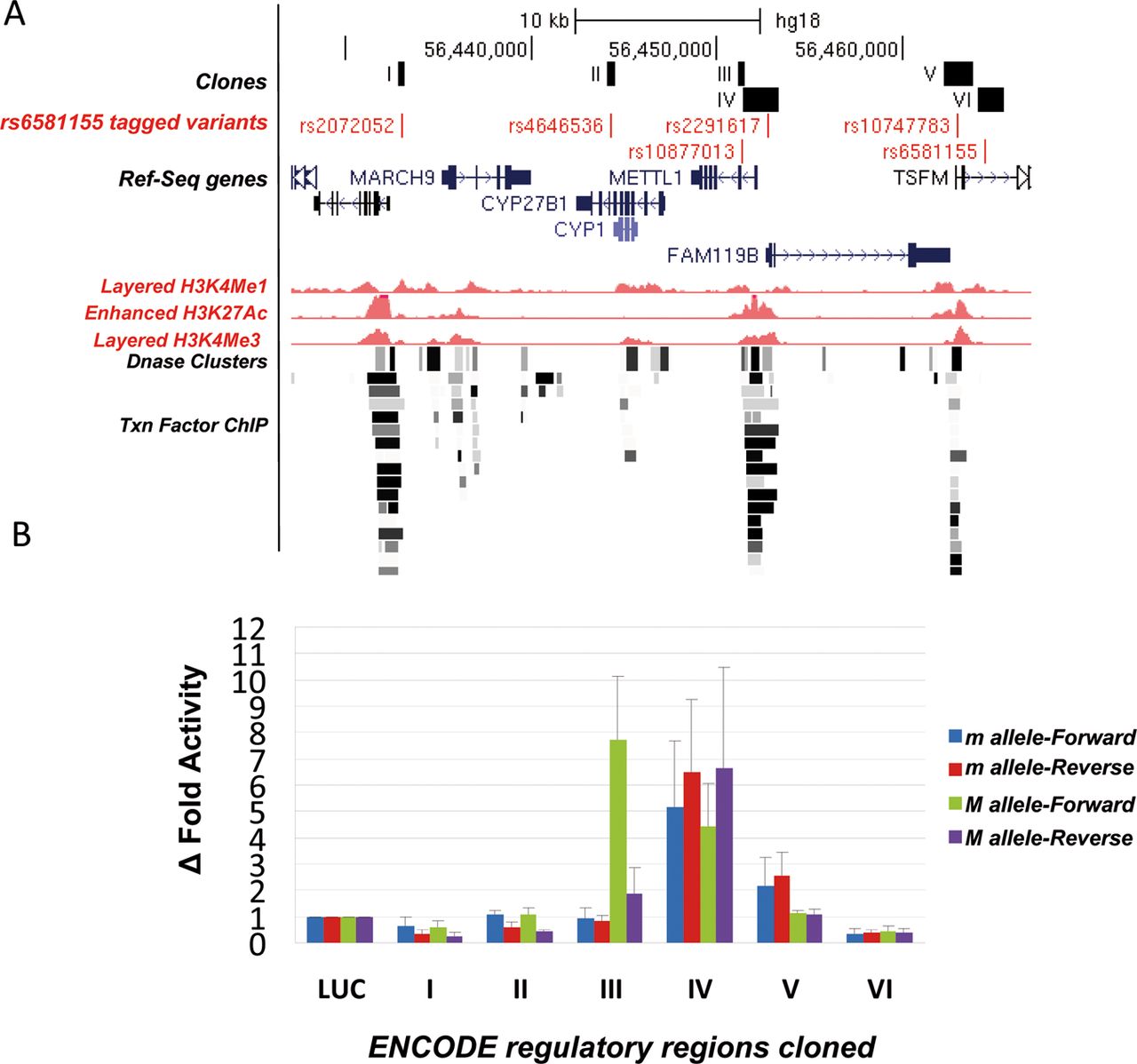
Since all the rs6581155-tagged polymorphisms were located in intronic and intergenic non-coding regions, another strategy to search for functionality was to use annotations of gene regulatory regions from ENCODE (http://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=173506627&c=chr8&g=wgEncodeReg). Six out of 18 captured SNPs were located in remarkable regions associated with histone methylation, binding sites of transcription factors or in DNAse I hypersensitive site as shown in figure 2A.

Enhancer-suppressor activity of the rs6581155-tagged variants. (A) Schematic illustration to show the localisation of the six tagged SNPs (in red) in potential regulatory regions (in black, I–VI) as indicated by ENCODE for the GM12878 lymphoblastoid cell line and the genes present in the region: Enhancer H3K4Me1 track shows where modification of histone proteins is suggestive of enhancer; Promoter H3K4Me3 track shows a histone mark associated with promoters; Layered H3K4Me3 track shows histone mark associated with promoters that are active or poised to be activated; DNase Clusters track shows regions where chromatin is hypersensitive to DNase I enzyme; Txn Factor ChIP track shows DNA regions where transcription factors bind to DNA as assayed by chromatin immunoprecipitation (ChIP) with antibodies specific to the transcription factor followed by sequencing of the precipitated DNA (ChIP-Seq). (B) Δ Fold luciferase activity respect to the empty vector of the different constructs corresponding to the six regions (I–VI) with potential regulatory activity, containing the rs6581155 tagged polymorphisms transfected into Raji B cells. Four clones for each region bearing the different alleles (allele m, minor and M, major) and in both orientations (Forward and Reverse) from three independent transfection experiments are represented. Luciferase activity levels are referred to the level of the control plasmid containing only the basic promoter and the Renilla activity.
To test their potential activities as enhancers or repressors of gene expression, we cloned the fragments containing these six variants upstream of the basic promoter of a luciferase reporter system and transfected into Raji lymphoblastic B cells.
We cloned four inserts for each of the six regions carrying both alleles in forward and reverse orientation. Only region III containing variant SNP rs10877013 had allele-dependent and orientation-dependent enhancer activity. Thus, allele A versus allele G in forward orientation showed 9.5-fold higher ratio of luciferase activity (figure 2B). We observed a 2.7 times higher enhancer activity of the T allele in the reverse fragment.
TFSEARCH, Match and TESS software were used to predict potential transcription factor binding sites that could be responsible of the different allele-depending activity of fragment III. The SNP rs10877013 falls within the putative binding sites for the CCAAT/enhancer binding proteins (C/EBP) α and β. Although both alleles appear to be consistent with the consensus sequence for C/EBP TF it is possible that a base change could affect the affinity or isotype binding of the C/EBP transcription factor to the sequence.
Discussion
The 12q13.3–12q14.1 region has been associated to several autoimmune diseases8 ,12–16 but no causal variant has been found so far for any of them exception made for a rare variant in the coding region of CYP27B1 gene associated with MS.17 This variant was not detected in our sample set.
In the present work, by integrating several strategies, we have uncovered the variant most strongly associated with MS in the region. This variant also affects the activity of an enhancer in an allele-dependent and orientation-dependent manner and correlates with the expression of five genes of the locus. Interestingly, the major allele of this SNP (rs10877013) correlates with high luciferase activity and at the same time with high expression of TSFM and TSPAN31 and low expression of FAM119B, CYP27B1 and AVIL. Although it could seem contradictory that one enhancer may differentially affect the expression of different genes, parallel situations have been described extensively in two scenarios: first, the same transcription factor is capable of activating or repressing depending on the context;32 ,33 second, a single DNA sequence can be bound by different factors with opposite activities.34 On the other hand, we cannot forget the important role of H3K4me preventing aberrant gene expression or modulating transcriptional response. H3K4me turns to mark for both positive and negative regulation. The role of H3K4me2 in histones deacetylation could serve as a fine-tuning regulatory mechanism.35
It has been described that interacting promotes are coexpressed in a tissue-specific manner. Based on the Chromatin Interaction Analysis with Paired-End-Tag sequencing results mapping long-range chromatin interactions associated with RNAPII and H3K4me2,36 ,37 we observed that the genes in the analysed region are engaged through promoter–promoter, promoter–enhancer interactions forming a multigene complex that could provide a structural framework for cotranscription (figure 3). In this sense, the effect of a polymorphism on one enhancer embedded in this multigene complex could have diverging effects depending on the interaction of this enhancer with the different promoters. Additionally, the interactions could be tissue-type or cell-type specific, as has been shown by Li et al.36 Therefore, it is not evident which of the genes in the region may be responsible for the genetic association with MS, since they seem to be coordinately regulated, and the enhancer containing the MS-associated variant may be affecting either of them; in fact, it acts as an eQTL for all of them. We observed that the Spearman's correlation coefficients (r) between the expression levels of the different genes in this cluster and the rs6581155-tagged SNPs were different in the different assays performed. For instance, in monocytes the best correlation is observed with FAM119B (r=−0.52), however in data obtained from LCL by RNA-seq a medium-high correlation coefficient is found for both CYP27B1 (r=−0.472) and FAM119B (r=−0.474), and with microarrays the correlation coefficient is very similar for TSFM and FAM119B. Based on data from LCL cells, TSFM, FAM119B or CYP27B1 would be candidates, while FAM119B appears to emerge from monocytes. In fact, Gandhi et al20 reported that FAM119B expression level was much lower in MS patients carrying the susceptibility haplotype. We observed that this correlation occurs also in samples from healthy donors as they are the CEU HapMap samples. However, we do not know the precise conditions (cell type, stage of disease, etc) under which expression of these genes could be determinant for the development of MS. For the same reason the variant-specific enhancer activity of fragment III, reported in this work, could vary in different cell-types. Here we have used in the activity assays the same cell-type used in the eQTL analysis to reproduce the effect observed in the expression of these genes. The Raji cells are lymphoblast-like cells derived from B-lymphocyte as the LCL from HapMap. The effect of the enhancer on the gene expression of proximal tubule cells of the kidney and the disease-activated macrophages, which are the major source of CYP27B1,38 could shed light on the involvement of this variant in MS.

{kind=link}
{kind=link}
{kind=link}
Datasets on Long-Range Chromatin Interaction with Paired-End-Tag sequencing (ChIA-PET) in the studied region. (A) Coordinates of chr12 in B37 and known genes by University of California Santa Cruz (UCSC) browser. (B) H3K4me2 ChIA-PET data obtained from CD4 T cells, data from Chepelev et al.37 (C) RNAPII ChIA-PET data obtained in K562 cells (data from Li et al36) In red, the number of the PET-counts (PETc) of the most frequent interactions is indicated. (D) RNAPII ChIA-PET data obtained in MCF7 cells.36 The most frequent interactions are indicated in red. (E) ENCODE information on transcription levels assayed by sequencing of polyadenylated RNA from K562 cells. (F) ENCODE information on the RNAPII binding sites by ChIP-seq. (G) H3K4Me3 histone mark in K562 cells associated with promoters. (H) H3K4Me1 modification of histone proteins in K562 cells, suggestive of enhancer and, to a lesser extent, other regulatory activity.
Relying on published data from other groups on the concentration of precursors of vitamin D in serum, two independent groups reported a correlation between the major alleles of rs703842, tagged by rs6581155 in the present work, with high 25-hydroxyvitamin D (25(OH)D) concentrations in MS patients.39 ,40 On the other hand, Lange et al41 reported a correlation of the major allele of rs10877012, in total LD with rs6581155, and low 1,25(OH)2D serum levels. The major allele of the MS-associated SNPs, in total LD with the previously mentioned SNPs, correlated with low expression of the CYP27B1 gene. This enzyme catalyses the conversion of 25(OH)D to 1,25(OH)2D, thus a low expression of this enzyme, could reconcile the above seemingly conflicting data, via accumulation of 25(OH)D and a low 1,25(OH)2D product. These data support the CYP27B1 gene as a very plausible candidate gene for association of the region with MS.
Vitamin D, besides having well-known control functions of calcium and phosphorus metabolism, bone formation and mineralisation, also has a complex role in the maintenance of immune homeostasis. The administration of vitamin D in animal models leads to improvement of immune-mediated symptoms. The correlation between the MS- associated variants and circulating levels of vitamin D supports the important role of vitamin D in susceptibility to MS. Thus, if this is the case in human MS, both the pharmacogenomics and the expression analysis of all the genes affecting the causing factor can reveal the kind of intervention necessary for neutralising a pathogenic situation.42
In this work we localise a group of variants in almost total LD that explain the association of the 12q13.3–12q14.1 region with MS. One of these variants (rs10877013) strongly affects the activity of one enhancer in the region, in an allele-dependent and orientation-dependent way, that could be the cause of the alteration of TSFM, TSPAN31, FAM119B, CYP27B1 and AVIL gene expression, due to the promoter multigene interactions observed in the region. Some of the tagged variants have been described to be associated with the vitamin D serum concentration. Our results point to one of these as the causal gene for MS association in the locus.
Acknowledgments
We thank patients with multiple sclerosis and control subjects for making this study feasible. SNP genotyping services were provided by the Spanish Centro Nacional de Genotipado CEGEN-USC (http://www.cegen.org).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online tables
Footnotes
AA, MF, OF, AS, EU and FM contributed equally.
-
Contributors AA, EU, KV and FM designed the study, performed the main statistical analyses and co-wrote the manuscript. MF carried part of the experiments. OF, AS, GI, ML, LL, JAGL, IA, AA, MJGB, JV, BH, RA, MC, XM, DO, JO, YB coordinated sample collection. MAA, NPM, AN carried out some bioinformatics analysis.
-
Funding Financial support for the study was provided by Ministerio de Economía y Competitividad (MINECO)-Fondos Europeos de Desarrollo Regional (FEDER) (grant number SAF2009-11491); Fondo de Investigación Sanitaria (FIS) (grant numbers RETICS-REEM RD07/0060, CP 10/00526, PI10/1985, PS09/02105); Junta de Andalucía- Fondos Europeos de Desarrollo Regional (FEDER) (grant numbers P07-CVI-02551, P09-CTS-5218); Ikerbasque; the Basque Foundation for Science (Bilbao) and Fundación Ilundain.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval This study was approved by the Institutional Review Boards of Hospital Regional Universitario Carlos Haya of Malaga, Hospital Clinic of Barcelona, Hospital Virgen Macarena of Sevilla, Hospital de Basurto of Bilbao, Hospital Clínico San Carlos of Madrid, Hospital Universitari Vall d´Hebron of Barcelona and Hospital Donostia of San Sebastian.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement The data presented in the manuscript are available on request.
-
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/