Article Text

Abstract
Background Bardet-Biedl syndrome (BBS) is a pleiotropic recessive disorder that belongs to the rapidly growing family of ciliopathies. It shares phenotypic traits with other ciliopathies, such as Alström syndrome (ALMS), nephronophthisis (NPHP) or Joubert syndrome. BBS mutations have been detected in 16 different genes (BBS1-BBS16) without clear genotype-to-phenotype correlation. This extensive genetic heterogeneity is a major concern for molecular diagnosis and genetic counselling. While various strategies have been recently proposed to optimise mutation detection, they either fail to detect mutations in a majority of patients or are time consuming and costly.
Method We tested a targeted exon-capture strategy coupled with multiplexing and high-throughput sequencing on 52 patients: 14 with known mutations as proof-of-principle and 38 with no previously detected mutation. Thirty genes were targeted in total including the 16 BBS genes, the 12 known NPHP genes, the single ALMS gene ALMS1 and the proposed modifier CCDC28B.
Results This strategy allowed the reliable detection of causative mutations (including homozygous/heterozygous exon deletions) in 68% of BBS patients without previous molecular diagnosis and in all proof-of-principle samples. Three probands carried homozygous truncating mutations in ALMS1 confirming the major phenotypic overlap between both disorders. The efficiency of detecting mutations in patients was positively correlated with their compliance with the classical BBS phenotype (mutations were identified in 81% of ‘classical’ BBS patients) suggesting that only a few true BBS genes remain to be identified. We illustrate some interpretation problems encountered due to the multiplicity of identified variants.
Conclusion This strategy is highly efficient and cost effective for diseases with high genetic heterogeneity, and guarantees a quality of coverage in coding sequences of target genes suited for diagnosis purposes.
- Targeted sequencing
- ciliopathies
- Bardet-Biedl syndrome
- multiplexing
- diagnosis
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
Bardet-Biedl syndrome (BBS; OMIM# 209900) is a pleiotropic recessive disorder with high non-allelic genetic heterogeneity. Its incidence varies from an estimated 1:160 000 in northern Europe to 1:13 500–17 000 in Bedouins and Newfoundlanders, respectively.1 BBS belongs to the large and growing family of ciliopathies and, therefore, shares phenotypic traits with Joubert (JBTS), Alström (ALMS) and Meckel (MKS) syndromes.1 ,2 Differential clinical diagnosis may thus be difficult, especially in young probands who do not yet show some later onset-specific manifestations.3 ,4 In particular, recent reports highlight a significant clinical overlap between BBS and ALMS.3 ,5
The main phenotypic features of BBS comprise retinal dystrophy, polydactyly, obesity, mild developmental delay, polycystic kidneys and hypogenitalism. Other minor features can also be observed in patients, such as cardiac abnormalities, other digit or eye anomalies, diabetes, hypertension, hearing defects, anosmia.6 ,7 Up to now, mutations have been detected in 16 different genes (BBS1-BBS16), but no clear genotype-to-phenotype correlation could be observed, besides the suggested exception of BBS16.8
Alström syndrome (OMIM #203800) was reported to be much less prevalent than BBS, with an estimated incidence of 1:1 000 000. Its phenotypic features overlap with those of BBS in early infancy and include: cone-rod dystrophy, obesity, type 2 diabetes mellitus, hearing loss but also hypertriglyceridemia, dilated cardiomyopathy, and progressive pulmonary, hepatic, or renal dysfunction.9 To date, only one gene (ALMS1) has been identified, but recent reports showed some families with suggestive ALMS-carrying mutations in BBS genes.3 ,5 The large size of ALMS1 coding sequence appears to have impaired widespread diagnostic testing of ALMS.
Exhaustive conventional Sanger sequencing for BBS diagnosis is prohibitively expensive because of the large number of genes involved, and so also for ALMS due to the large size of ALMS1 coding sequence (12 kb, 24 exons; table 1). Alternative cost-conscious strategies have been proposed for BBS diagnosis, such as: initial screening of recurrent mutations and frequently mutated genes (BBS1, BBS10, BBS12) combined with homozygosity mapping for consanguineous families10 ,11; or primer extension arrays to test a series of known BBS mutations.5 Another approach recently proposed is the pooling of patients' DNAs with subsequent PCR-amplification and massive parallel resequencing of BBS1-12 coding exons, followed by heteroduplex screening to identify the mutation carrier.12 Such a method presents some limitations as it will miss exon deletions and may not be suited for diagnostic purposes. Considering the clinical overlap with other ciliopathies, another approach would be to test, systematically and simultaneously, all corresponding genes for such overlapping syndromes, which would be particularly relevant for patients with atypical or incomplete clinical phenotypes. We describe here the results of such an approach, based on a targeted exon capture of 30 genes coupled to next-generation sequencing (NGS).
Genes included in the targeted enrichment strategy and their associated disorders
Subjects and methods
Detailed protocols are available in Supplementary Methods.
Subjects
DNA samples from 52 unrelated patients were collected. Most patients had been addressed to the diagnostic laboratory, or to the National Reference Center for rare ophtalmogenetic diseases in Strasbourg. Eleven DNA samples stemmed from Tunisian patients included in an independent BBS epidemiology study.
The proof-of-principle cohort included 14 non-Tunisian patients with a confirmed BBS molecular diagnosis (identified prior to this study by Sanger sequencing). Twenty-six out of the 38 patients without known mutations, and recruited in Strasbourg, had been initially screened for BBS1 and BBS10 recurrent mutations, plus the entire coding sequence of BBS12.
For all patients, a written consent for genetic testing was obtained, either from adult probands or from the legal representative in case of minors.
Library preparation, targeted capture and sequencing
DNA samples were prepared and controlled following standard procedures.
The capture design was performed with eArray following the manufacturer's instructions (Agilent).
DNAs (3 μg) were sheared mechanically using Covaris E220 (duty cycle: 10%; intensity: 5; cycles per burst: 200; time: 300 s).
For the proof-of-principle experiment, sequencing adaptors were added on 500 ng of sheared DNA using the SPRIworks Fragment Library System I (Beckman Coulter). After amplification and quality assessments, targeted capture was performed on individual samples using the in-solution SureSelect Target Enrichment System (Agilent) on 500 ng of DNA-prepped library. Additional steps of washing, purification and elution were performed, and multiplexing adaptors (TruSeq Illumina DNA indexes) were added by PCR during the post-capture amplification step.
For all following experiments, multiplexing adapters were added simultaneously to sequencing adapters using the SPRIworks system. Equimolar amounts of two tagged libraries were then pooled prior to the capture reaction. All other following steps prior to sequencing remained identical. A 72-bp single-read sequencing was performed on a Genome Analyser IIx (GAIIx, Illumina).
Bioinformatic pipeline
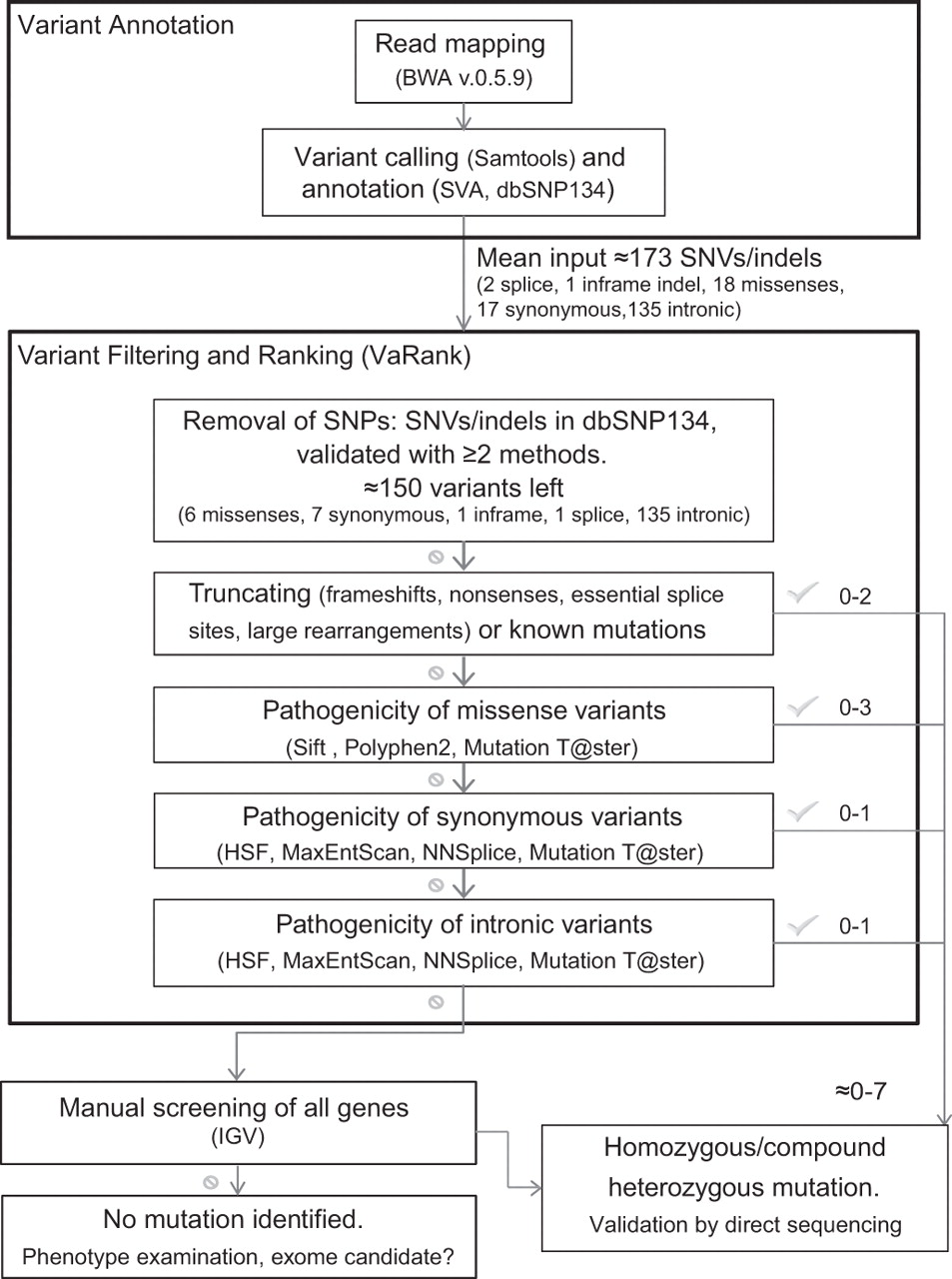
Read mapping and variant calling were performed following standard procedures. Variant filtering was performed using VaRank, an in-house software which collects variant-specific information to rank them according to their predicted pathogenicity (figure 1, Supplementary Methods).

Global flowchart of the bioinformatic pipeline implemented for mutation detection. Software acronyms: BWA, Burrows-Wheeler Aligner; SVA, Sequence Variant Analyser; SIFT; Polyphen2; HSF, Human Splicing Finder; MaxEntScan, Maximum Entropy Scanning; NNSplice; Mutation Taster; IGV, Integrative Genome Viewer.
Copy-number variation (CNV) detection method
CNVs were identified using a depth-of-coverage method.13 ,14 For each patient, read counts in non-overlapping windows of 20 nucleotides were computed, normalised and then compared randomly with eight other samples from the same experiment (considered as replicates) using the Bioconductor package DEseq (initially designed for RNA-seq data).15 Candidate regions for CNVs were retrieved when log2 ratios (controls/sample) were either ≥0.84 (fold change >1.8, potential deletion) or ≤−0.51 (fold change <0.7, potential duplication), and if p values adjusted for multiple testing (Benjamini and Hochberg procedure)16 were smaller than 0.1.
Statistical methods
Confidence intervals were computed for proportion estimates and indicated in brackets. Fisher's exact test was computed to compare distributions of small populations. Subsequent p value is given at α=0.05.
Results and discussion
Targeted regions: design strategy
Our primary goal was to develop an efficient mutation-screening strategy for the diagnosis of patients with phenotypes evocative of BBS, or of clinically overlapping ciliopathies. We chose a target enrichment approach coupled with NGS in order to focus the sequencing on genomic regions of interest. We targeted all exons (including 5′ and 3′ UTRs) of the 16 known BBS genes (table 1). Because of the known clinical overlap, we also included coding exons of ALMS1, and of all 12 known nephronophthisis genes (NPHP1-12), since retinal degeneration can often be observed in this kidney-specific disease.9 ,17 Coding sequences of AHI1/JBTS3, TMEM216/MKS2/JBTS2, and of the proposed BBS-modifier CCDC28B/MGC1203, were also targeted.18 Because some of these genes are associated with multiple phenotypes, our design includes 6 MKS, 7 JBTS and 4 Senior-Loken syndrome (SLSN) genes (see table 1).
With this first design, we wanted to investigate whether including intronic sequences could favour both, the detection and sizing of exon deletions. We therefore included baits-targeting intronic sequences of BBS1 and BBS4. This choice was dictated by two observations: an apparent excess of patients heterozygous for the BBS1-recurrent mutation M390R with no second mutation detected,11 and multiple reports of BBS4 exon deletions in patients.4 ,11 ,19 A maximal threshold of 200 kb for cumulated targeted regions was set because of the manufacturer's pricing limits.
Presence of repeated sequences precluded bait tiling in 19.7% of initially targeted regions. This concerned, almost exclusively, introns of BBS1 and BBS4, besides a small number of 3′UTRs, and only 128 bp of protein coding regions (within first exons of ALMS1 and NPHP3; table S1).
Proof-of-principle and technical results
In our proof-of-principle experiment, we selected 16 DNA samples, of which 14 were with known BBS mutations. In this first trial, after barcoding the target-enriched libraries, we sequenced pools of four or eight libraries per lane of a GAIIx (see Supplementary Methods). This proof-of-principle analysis was carried blind, that is, without knowledge of implicated BBS genes and their associated mutations. A constellation of all mutation types (missenses, nonsenses, splice mutations, large deletions and complex rearrangements) at different allelic dosage was tested (figure S1). All 14 previously identified mutations, including two heterozygous BBS1-deletions (figure 2A), were detected in their correct heterozygous/homozygous state (table S2). In particular, in patient AKE12, we could detect an abnormal local drop of coverage in BBS12 due to a rare mutation type (insertion of an Alu sequence, figure S1A) although the exact nature of the mutation could only be determined by Sanger sequencing. A similar drop in coverage was observed for a second patient, AHX91, with another complex mutation detected previously by Sanger sequencing (insertion/inversion in BBS5).
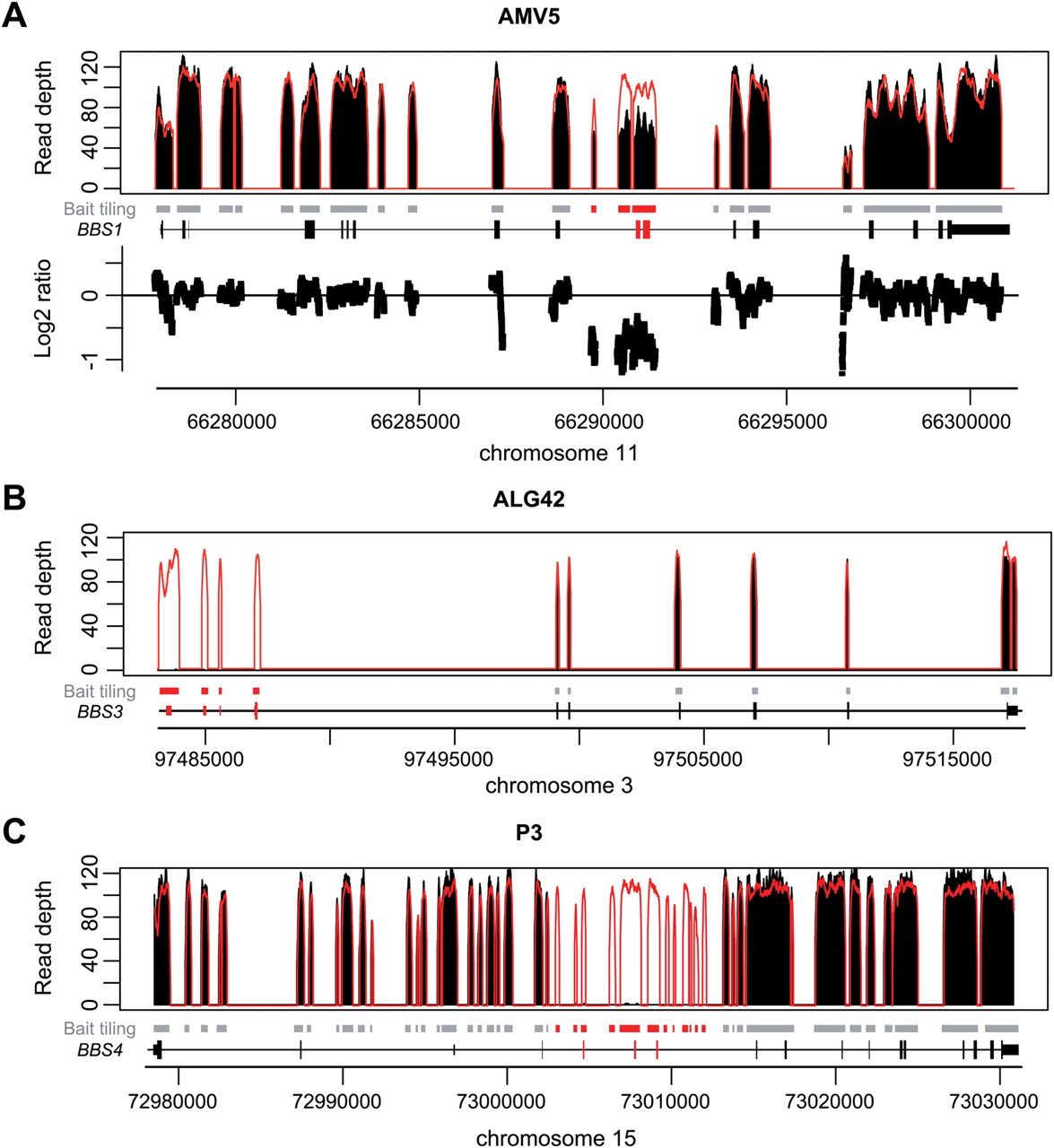
Detection of large deletions in three patients using a depth-of-coverage method. Black peaks: normalized depth of coverage from patients' DNA samples. Empty peaks: normalized mean depth of coverage across samples from the same sequencing lane. Grey squares: bait-covered regions. Black peaks: normalized depth of coverage from patients' DNA samples. Empty peaks: normalized mean depth of coverage across samples from the same sequencing lane. Grey squares: bait-covered regions. Highlighted squares: deleted regions. Gene representation: black squares: exons, dashed lines: introns. Genomic positions are given according to the human reference genome hg19/ GRCh37. (A) Heterozygous deletion of BBS1 (exon #10, 11) in AMV5 patient. Corresponding Log2 ratios between both depths of coverage (normalised mean and AMV5 patient) further highlight the presence of the deletion. (B) Homozygous deletion of BBS3 (exon #1, 2a, 2b, 3) in ALG42 patient. (C) Homozygous deletion of BBS4 (exon #4, 5, 6) in P3 patient. (A and C): targeting intronic sequences allows restricting the deletion breakpoints. (B) and C): Log2 ratios between both depths of coverage (normalised mean and corresponding patients) could also allow detecting both deletions but are not shown (supplementary figure S2).
In this first experiment, we almost systematically reached the maximal theoretical coverage of 144x illustrated by a mean coverage of 127±4x after removal of duplicate reads (table 2). Due to this global saturating coverage when considering unique reads, we used all reads, including duplicates, when applying our depth-based method for the detection of CNVs.
Sequencing statistics of both coverage (in captured regions) and capture efficiency
These promising depth-of-coverage results (table 2, table S3) encouraged us to further increase the number of pooled samples. In the next experiments we used a single capture reaction for two barcoded libraries, allowing both cost and bench-time savings, and pooled 12 libraries per sequencing lane (maximum number of barcodes proposed at that time by Illumina).
This new protocol was performed on a second cohort of 36 patients with unknown mutations. Sequencing resulted in a mean coverage of 78±17× (283±153x before discarding duplicate reads) with 91.4±6.4% of targeted regions being covered more than 40x (table 2). This relative drop of coverage appears to be a consequence of a lower capture efficiency that might be due to: (1) an input amount of individual library reduced by half, due to the pre-capture pooling and (2) the addition of barcodes before capture, leading to less efficient blocking and unspecific hybridisation. The resulting coverage still guarantees a reliable detection of variants and of their homozygous/heterozygous state.
A small proportion of targeted regions was weakly covered in some patients (ie, depth <10x after duplicates filtering), with very few of them in a systematic way in other patients (table S4). This only concerned 0.63±0.68% of protein coding regions, and mostly included intronic GC-rich sequences (GC content: 68.3±5% vs 40.2±10% across all targeted regions), or some first exons (tables S3 and S4).
Variant filtering: importance of databases and frequency data
In targeted regions, we detected, on average, 170 variants (Single Nucleotide Variants (SNVs) and indels) per patient. All were systematically analysed for putative effect on protein structure and splice sites using VaRank (figure 1, Supplementary Methods). About 130 of these variants were recorded in dbSNP134 (table S5), but only 20 were validated with at least two independent methods and, therefore, filtered out. Indeed, in the context of a rare recessive disorder, some true mutations can be present at very low frequency in a heterozygous state in controls.
Potential pathogenicity of the remaining 150 variants was assessed using bioinformatic tools and considering their allele frequency in a European-American population, as reported in the Exome Variant Server database (EVSdb). This yielded from zero to six interesting variants per sample, among which were obvious truncating or known mutations in some patients.
The new ‘clinical significance’ field introduced in dbSNP134 has to be considered with caution since established mutations can now be reported in the database but are not systematically flagged as pathogenic (example: rs179363897, p.R138H) mutation in BBS5). Conversely, we detected some false-positive annotations: rs4784677 (p.N70S) in BBS2—initially reported as a third allele according to the triallelic hypothesis—20 is flagged as pathogenic, but is too frequent to be a fully penetrant mutation (0.77% in EVSdb). Filters have to be carefully adapted to the disorder of interest, and to the constantly evolving updates of databases.
Detection of exon deletions
One advantage of NGS-based strategies, as opposed to Sanger sequencing, is the opportunity to detect—in addition to SNVs and small indels—CNVs affecting one or more exons (figure 2 and S2). In the proof-of-principle experiment, two heterozygous deletions could be detected in BBS1. Among the unknown samples, two homozygous deletions in BBS3/ARL6 and BBS4 were identified. To our knowledge, we provide here the first report of large deletions in BBS1 and BBS3/ARL6, while several deletions affecting BBS4 have been previously observed.4 ,11 ,19 Since we also targeted intronic sequences of BBS1 and BBS4, we were able to narrow the boundaries of subsequent detected deletions (figure 2). For patient AMV5, by using coordinates of affected exons, the estimated size of BBS1 deletion would be between 466 and 4707 bp, while with our design, we could restrict it to 1862–3841 bp (figure 2A, table S2). In patient P3, we could similarly reduce the assessed size of the BBS4 deletion from 4626–12 975 bp based on exon positions down to 9376–10 469 bp (figure 2C, table 3A). Lastly, since the BBS3/ARL6 deletion in patient ALG42 encompasses the first three exons of the gene (figure 2B), we tested whether it may extend and affect EPHA6 located upstream, encoding an ephrin receptor. Direct PCR testing excluded such extended deletion (data not shown).
Identified mutations and other potentially pathogenic variants in the 38 patients with previously unknown genotype. A) Patients with two clearly pathogenic variants in one gene; B) Patients with a single or no clear pathogenic variant in one gene. Mutations are described according to the latest nomenclature conventions described in HGVS
Thanks to this method, in the six patients in whom we detected a single heterozygous potentially pathogenic mutation, we can ascertain that no heterozygous deletion is present in trans, or at least none encompassing exonic sequences.
Distribution of detected BBS mutations in the 38 unknown patients
Of the 38 samples with unknown BBS mutations (36+2 from the proof-of-principle experiment) we detected clearly pathogenic biallelic mutations in 26 cases (68.4%; table 3A). To our knowledge, seventeen of these mutations have not been reported previously, indicating that the BBS mutation spectrum is far from being saturated in spite of numerous BBS mutation reports. Homozygous mutations were found in 88% (23/26) of mutated patients, coherent with the large number of consanguineous probands included in the cohort (75%; 25/33). In two patients of consanguineous origin, the BBS mutation was located outside the homozygous regions detected by prior SNP array analysis and would, therefore, have been missed using a homozygosity mapping strategy (patients AGL23, AKX44; table 3A).
Among the remaining 12 patients with no biallelic mutations identified (table 3B), one patient (AHR2) had a heterozygous clearly pathogenic splicing mutation in BBS3. Five patients had heterozygous missenses predicted to be damaging by SIFT, Polyphen2 and/or Mutation T@ster (AIY87, AIX45, AMO77, AMA28, AHL86) with the latter two carrying such variants in two different genes. One consanguineous Melanesian patient, AKE98, presented with classical BBS-inclusion features, including polydactyly. He carried two homozygous variants which initially appeared as potentially pathogenic: a distal frameshift in INVS/NPHP2 predicted to add 14 amino-acids to the C-terminus of the longer protein isoform, and a non-reported missense P2679L affecting a conserved residue in ALMS1 (figure S3). Subsequent segregation analysis ruled out their implication in the disease since both variants were heterozygous in a similarly affected brother.
In five patients, no potentially pathogenic variant could be identified in any of the 30 targeted genes. These patients are thus candidate for exome sequencing which might either help in identifying novel genes or in reconsidering the clinical diagnosis.
Mutation load in BBS and other targeted genes: importance of ALMS1
The mutation load among BBS genes in our cohort appears consistent with previous reports.7 ,11 Observed occurrences for BBS1 (7/38, 18.4%) and BBS2 mutations (5/38, 13.2%), the most frequently mutated genes in our study, are similar to the respective reported figures of 16.9% and 12%.7 Considering frequently mutated genes, our study was strongly biased against BBS1, BBS10 and BBS12, since two-thirds of the patients' DNAs were previously tested negative for BBS1 and BBS10 recurrent mutations, plus all BBS12 protein coding sequence. This explains the total absence of BBS12 mutations in our cohort, and the relatively low contribution of BBS10 (5/38, 13.2%) as compared with the literature (≥20%).7 ,11 The contribution of other BBS genes was low, with frequently only one proband involved.
We did not find any mutation in the ‘new’ BBS genes (BBS13–16) suggesting that, cumulatively, they have a small contribution to the total mutation load. BBS13/MKS1 was indeed shown to be mostly implicated in MKS since only one BBS patient was reported with two heterozygous mutations p.[C492W]; [F371del] others carried only heterozygous missenses, sometimes in addition to homozygous truncating BBS1 mutations.21 Likewise, for BBS14/CEP290, a homozygous truncating mutation (p.E1903*) was found in a single BBS patient,21 while other mutations are much more often implicated in Joubert, Senior Loken, Leber Congenital Amaurosis or Meckel syndromes. Similar observations can be made for BBS15/WDPCP and BBS16/SDCCAG8.22 ,23 Like in all other studies of BBS cohorts, no mutation was identified in BBS11/TRIM32 raising the question of its real implication in BBS: only one homozygous missense mutation was described in a single consanguineous family, while several other mutations were identified in recessive forms of limb girdle muscular dystrophy.24
One noteworthy result is the finding of homozygous truncating ALMS1 mutations in 3/38 patients (AIA84, AKO26, ALB64; 7.9%). In particular, the nonsense found in AKO26 patient p.R3629* seems to be a recurrent ALMS1 mutation, since already reported in five other ALMS patients.25–27 The phenotypic overlap between BBS and ALMS seems to be larger than previously thought, as recently suggested with examples of Alström patients with mutations in BBS genes,5 and the reverse situation, such as in our study, of ALMS1 mutations in patient with suspected BBS.3
Lastly, no clearly pathogenic mutation was found in any NPHP or JBTS genes in the cohort.
Correlation between mutation detection efficiency and clinical phenotype
Comparison of clinical phenotype between patients with two clearly pathogenic mutated alleles (n=26) and those with either a single possible pathogenic variant or no suspicious variant detected (n=12) showed a clear correlation between the number of major BBS clinical features and the probability of detecting two BBS mutated alleles in patients (figure 3). Biallelic mutations were detected in 81% (CI (60% to 92%)) of patients meeting BBS inclusion criteria. In particular, in Tunisian patients recruited upon strict clinical criteria, mutations were found in 11/11 cases and in seven different BBS genes, ruling out a potential founder effect. On the contrary, for some of the 12 patients without clear mutations, BBS was only one suspected diagnosis among others. Furthermore, our initial selection of patients without recurrent mutations in BBS1 or BBS10, and without any mutation in BBS12 may have enriched our cohort in patients with non-typical BBS phenotypes. The current widely quoted estimation that known BBS genes account for only 70–75% of the total mutation load in BBS patients may thus be underestimated if considering only patients with strictly defined BBS phenotype.

{kind=link}
{kind=link}
{kind=link}
Compliance with classical BBS phenotype is positively correlated to the efficiency to detect principal mutations in BBS genes. (A) The number of BBS diagnostic major inclusion criteria6 in patients is correlated to an efficient detection of BBS mutations. (B) Efficiency of detecting mutation in patients fulfilling BBS the phenotypic inclusion criteria or not. BBS inclusion criteria presenting with three major features plus at least two minors, or presenting with four major features and more.6 Primary criteria include: rod-cone dystrophy, polydactyly, obesity, learning disabilities, hypogonadism and renal anomalies. Secondary features comprise speech delay, other eye anomalies, brachydactyly or syndactyly, ataxia, diabetes, developmental delay, dental anomalies, cardiac anomalies and hepatic fibrosis.6
The distribution of BBS inclusion features appears different between patients with two BBS mutations, two ALMS1 mutations or no biallelic mutation identified (table 4). Patients with no detected mutation presented with significantly less polydactyly, a major BBS clinical sign: only 25% versus 70% in patients with detected BBS mutations (p=0.029*). The other clinical features seem to follow the trend of classical BBS patients.
Report of major BBS clinical features in the 38 patients without previously known molecular diagnosis, with or without detected mutations
Regarding ALMS1-mutated probands, 2/3 had been sent for suspected BBS (Prader-Willi or ALMS were also considered for AIA84 and AKO26, respectively) and satisfied BBS diagnostic criteria; the last one (ALB64) was addressed for syndromic retinal dystrophy (table 4).6 AIA84 presents a classical BBS with retinal dystrophy, obesity, cognitive defects, hypogonadism and brachydactyly. AKO26 presents an atypical BBS with the same features along with abnormal severe deafness, specific for ALMS. Lastly, ALB64 presents a typical ALMS with severe deafness and retinal dystrophy. None of them presented with polydactyly. As previously suggested,3 both, the absence of polydactyly and the prevalence of deafness in ALMS1-mutated patients, are keys for genotype-phenotype discrimination between ALMS and BBS mutated patients.
Assessment of oligogenism in BBS
The presence and potential effect of triallelism or oligogenism in BBS has been widely discussed and appears controversial (18 ,20 ,28 ,29 vs30–32). In our approach, the simultaneous sequencing of all 16 BBS genes, and of 14 other genes involved in overlapping ciliopathies, allows the systematic detection of most additional potentially pathogenic variants in those genes and, consequently, an unbiased assessment of oligogenism.
Out of the 52 patients analysed, we found only one heterozygous truncating mutation (p.K1870Nfs*4) as a third allele in BBS14/CEP290 in patient AKR68 who carries a pathogenic missense mutation in BBS10. Such a frequency is in fact in the range of what to expect by chance. We previously calculated the probability of carrying a true BBS-pathogenic mutation to be about 1:50.31 Since we also included in our design other ciliopathy genes, the probability to carry a pathogenic mutation in one of the 30 genes is rather between 1:20 and 1:30 (calculation based on each disease incidence and reported contributions of targeted genes in the mutation load). Potentially, pathogenic heterozygous missenses (not previously reported in patients or in the EVSdb) were also found in eight patients (three of the proof-of-principle, table S2; five of the ‘unknown’ cohort, table 3). Such variants might act as modifiers, but it is unlikely that they are required for full expression of a classical BBS phenotype. Conversely, in some patients where a single clearly pathogenic mutation was found, variants in other genes of the same pathway (especially those encoding proteins of the same complex, such as the BBSome or the BBS-chaperonin complex)33 ,34 might contribute to the disease state in a digenic mode of inheritance proposed in few BBS families.20 ,35 ,36
Potential case of triallelism is illustrated by patient AIZ62, who is compound heterozygous or a nonsense p.E191* and a missense p.A242S in BBS6/MKKS. The pathogenicity of A242S variant has been a subject of discussion.37–39 Analysis in zebrafish indicated that it affects BBS6/MKKS function and suggested a dominant negative effect.40 EVSdb allows to infer its frequency at 0.59% (CI (0.43 to 0.80%)), higher than the most frequent BBS mutation, M390R in BBS1 (0.24%, CI (0.15 to 0.39%)). A242S cannot thus be a highly penetrant mutation since it should then be found more frequently in patients than the M390R mutation, which is not the case. In patient AIZ62, a third heterozygous variant was identified in BBS12 (p.Q620R, residue conserved in mammals, but not in more distant vertebrates) thus affecting another subunit of the BBS-chaperonin complex.34 We suggest that A242S is a hypomorphic allele that may lead to a phenotype when in trans, with a complete null mutation, and could be further potentiated by a hypomorphic allele affecting another subunit of the same complex. Segregation analysis in AIZ62 family could not be performed to test this hypothesis.
Lastly, we looked in our cohort for the allelic frequency of the previously proposed BBS-modifier variant c.330C→T in CCDC28B/MGC1203,18 and found a frequency of 3.85% (CI (1.56 to 9.47%)), which is not significantly different (p=0.17) from the 2.07% (CI (1.76 to 2.43%)) observed in EVSdb.
Conclusions
The extensive non-allelic genetic heterogeneity of Bardet-Biedl syndrome has been a major problem for molecular diagnostic and genetic counselling applications. Various strategies have been proposed in recent years to optimise mutation detection,5 ,10–12 but have either low sensitivity or are too time consuming and expensive in diagnostic settings. This problem is shared by other disease entities, such as cardiomyopathies, hearing loss, Usher syndrome or Charcot-Marie tooth neuropathies. Those NGS-based alternative strategies can be divided in whole genome/exome sequencing,41–43 or targeted sequencing, either by using multiplex PCR,12 multiple singleplex PCR44–46 or capture enrichment approaches.
We implemented an in-solution targeted capture strategy for the 16 known BBS genes and 14 other genes implicated in ciliopathies that share overlapping clinical features with BBS. We show here that this is very efficient since in a single sequencing lane of an Illumina GAIIx one can simultaneously analyse more than 99.4% of targeted protein coding sequences of these genes in 12 patients, with sufficient coverage to guarantee reliable detection of heterozygous variants, small indels and exon deletions. In a week-long run, one could thus potentially analyse 96 patients. Investigation of 36 patients could be completed in about 3 weeks when including sample preparation and initial bioinformatic analysis. Data analysis and further mutation validation takes a longer time for cases where only variants of uncertain pathogenicity are present. We estimated the overall consumable costs in our settings at about $600. This can be even further decreased by using the latest more powerful sequencers, allowing analysis of larger pools of barcoded samples while keeping a high depth of coverage. Indeed, we recently sequenced a new cohort of 24 patients (using 12 capture reactions) in a single lane of an Illumina HiSeq2000 with even higher coverage than the one obtained in previous experiments with the GAIIx (data not shown). While exome sequencing is clearly more exhaustive, in terms of gene coverage in current implementations, 20–30% of targeted exons are not sufficiently covered for diagnostic accuracy, that is, to ensure low rates of false-positive/false-negative findings and reliable detection of heterozygous mutations or exon deletions. Moreover, the informatics resources needed for exome/genome sequencing data analysis and storage are considerably more important than for targeted sequencing, and can often be a limitation.
This strategy, however, presents some limited pitfalls. Few protein coding regions were not well covered, either because of failure in bait design (presence of repeat motifs) or poor capture efficiency (mostly GC-rich sequences and first exons). Then, our protocol with initial barcoding of libraries followed by capture on pooled samples may be cost effective, but at present, limits the capture efficiency and needs further optimisation, especially if applied to larger gene sets. Finally, like for all targeted exon strategies, deep intronic mutations will be missed. The alternative to targeting entire genes would still miss a high proportion of intronic regions containing repetitive sequences, and would also disproportionately increase the number of rare variants to analyse with splice-prediction bioinformatic tools that are currently not highly reliable. For genes expressed in leucocytes or fibroblasts, another alternative would be selected RNA sequencing (enriched in cognate gene transcripts).
While similar capture strategies have been recently developed for other diseases, most of them included a much smaller cohort, and reported only proof-of-principle analysis,47–49 with the exception of Walsh et al.50 Multiplex PCR approaches may have the potential of covering exons more exhaustively,46 given that primer design is more flexible than hybridisation bait tiling, but is limited to smaller gene panels than for exon capture. PCR pooling without barcoding has been used for BBS and NPHP,12 ,51 a strategy which may be cost effective for analysing large numbers of samples in epidemiological studies, but appears unsuited for diagnosis where a key preoccupation is to limit false-positive/negative rates.
Regarding the specific case of BBS, our study suggests that when strict clinical criteria are complied with, the frequency of detected mutations is higher than the generally quoted 70% figure.7 ,11 There may thus be only few strict BBS genes remaining to be identified, especially considering that with most strategies, with the exception of whole-gene sequencing, one will miss deep intronic pathogenic mutations. Additional genes to be discovered may correspond rather to variant BBS-like phenotypes than to strictly defined BBS. Indeed, for patients with BBS16/SDCCAG8 mutations, genotype-phenotype analysis showed for the first time a clear departure from the typical BBS phenotype with absence of polydactyly and systematic and severe renal manifestations (usually present in only 30% of BBS patients).7 ,8 Our finding of ALMS1 mutations in three patients confirms the major clinical overlap with BBS. Finally, we found no evidence for triallelism in our cohort of BBS patients.
Web resources
-
The URLs for online tools and data presented herein are:
-
OMIM: http://www.omim/org/e-Array (Agilent Technologies): https://earray.chem.agilent.com/earray/UCSC: http://genome.ucsc.edu/SVA: www.svaproject.org/
-
dbSNP: http://www.ncbi.nlm.nih.gov/projects/SNP/Mutation Nomenclature: http://www.hgvs.org/mutnomen/recs.htmlExome Variant Server, NHLBI Exome Sequencing Project (ESP): http://evs.gs.washington.edu/EVS/Integrative Genomics Viewer (IGV): http://www.broadinstitute.org/igv/Polyphen 2: http://genetics.bwh.harvard.edu/pph2/Sift Human Protein: http://sift.jcvi.org/www/SIFT_enst_submit.htmlMutation taster: http://www.mutationtaster.org/
Acknowledgments
We thank Ngoc-Hoan Nguyen for his help in the bioinformatics setup, and Cécile Pizot for the development of VaRank. We warmly thank Géraldine Greff, Anne-Sophie Jaeger, Manuela Antin, Elisabeth Scherrer, Serge Vicaire and Muriel Philipps for their technical assistance. Lastly, we wish to thank all patients and families included in this study, and our clinician colleagues who addressed patients for diagnostic analysis and provided clinical information, in particular: Drs P Abou-Jaoudé, C Baumann, H Flodrops, T Frébourg, D Genevieve, A Goldenberg, B Leheup, P Parent, P Petitjean, C Poitou-Bernert and S Taque.
References
Supplementary materials
Data Supplement
Files in this Data Supplement:
Footnotes
Funding This work was partially supported by a grant from Agence de Biomédecine to JLM and JM, by funds from APLM and by the Association Française contre les Myopathies (AFM) thanks to its support to the IGBMC sequencing platform.
Competing interests None.
Patient consent For children, consent was signed by the parents.
Provenance and peer review Not commissioned; externally peer reviewed.