Article Text

Abstract
Background There is considerable interest in the use of next-generation sequencing to help diagnose unidentified genetic conditions, but it is difficult to predict the success rate in a clinical setting that includes patients with a broad range of phenotypic presentations.
Methods The authors present a pilot programme of whole-exome sequencing on 12 patients with unexplained and apparent genetic conditions, along with their unaffected parents. Unlike many previous studies, the authors did not seek patients with similar phenotypes, but rather enrolled any undiagnosed proband with an apparent genetic condition when predetermined criteria were met.
Results This undertaking resulted in a likely genetic diagnosis in 6 of the 12 probands, including the identification of apparently causal mutations in four genes known to cause Mendelian disease (TCF4, EFTUD2, SCN2A and SMAD4) and one gene related to known Mendelian disease genes (NGLY1). Of particular interest is that at the time of this study, EFTUD2 was not yet known as a Mendelian disease gene but was nominated as a likely cause based on the observation of de novo mutations in two unrelated probands. In a seventh case with multiple disparate clinical features, the authors were able to identify homozygous mutations in EFEMP1 as a likely cause for macular degeneration (though likely not for other features).
Conclusions This study provides evidence that next-generation sequencing can have high success rates in a clinical setting, but also highlights key challenges. It further suggests that the presentation of known Mendelian conditions may be considerably broader than currently recognised.
- Exome sequencing
- unidentified genetic conditions
- medical genetics
- paediatrics
- clinical genetics
- complex traits
- genetic screening/counselling
- genetics
- genome-wide
- psychotic disorders (including schizophrenia)
- molecular genetics
- gastroenterology
- immunology (including allergy).
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Exome sequencing
- unidentified genetic conditions
- medical genetics
- paediatrics
- clinical genetics
- complex traits
- genetic screening/counselling
- genetics
- genome-wide
- psychotic disorders (including schizophrenia)
- molecular genetics
- gastroenterology
- immunology (including allergy).
Introduction
Whole-genome and whole-exome sequencing have proven remarkably successful in identifying the causes of Mendelian diseases. These analyses have generally depended on the availability of more than one unrelated affected individual and/or linkage evidence in at least one family. However, next-generation sequencing (NGS) has also succeeded in identifying causes of genetic conditions even when they are seen in only a single patient.1–3
Consequently, there is growing interest in the introduction of NGS into the clinic to aid in the diagnosis of conditions for which no genetic cause can be found with targeted testing or chromosomal arrays. However, in a clinical setting, patients with undiagnosed genetic conditions tend to present with a wide range of clinical features, and it is often necessary to consider each patient's genome individually, rather than looking for common disrupted genes in multiple cases with a similar phenotype. It is not clear what success rate NGS approaches will achieve in providing genetic diagnoses in this more challenging setting. In this study, we have evaluated the use of NGS to provide genetic diagnoses using 12 parent-child trios in which the child had congenital anomalies and/or intellectual disabilities due to unexplained conditions presumed to be genetic. Importantly, the patients were chosen to be representative of a clinical sample of undiagnosed genetic conditions, in that they were not selected for genetic tractability or phenotypic homogeneity.
Methods
Exome sequencing was performed on each patient and both parents using the Illumina HiSeq2000 platform and the Agilent SureSelect Human All Exon 50Mb Kit. Detailed methods for laboratory work can be found in the online supplementary methods.
Study population
The research protocol was approved by the Duke Institutional Review Board, and all human participants or their guardians gave written informed consent. Twelve families (child, mother and father) were recruited through the genetics clinic at Duke University Medical Center based on whether their child met two or more of the following criteria: (1) unexplained intellectual disability and/or developmental delay; (2) one major congenital anomaly; (3) 2–3 minor congenital anomalies; and (4) facial dysmorphisms. In addition, the families were required to meet the following eligibility requirements: (1) both biological parents available for testing; (2) previous clinically indicated genetic testing, including a chromosomal microarray (Affymetrix 6.0, http://www.affymetrix.com), had been normal; and (3) no evidence of effects of teratogens, birth asphyxia or non-accidental trauma. Subjects were not eligible if the mother was pregnant at the time of enrolment. Finally, results were only returned to patients and/or patient families following confirmation of detected variants in a CLIA certified laboratory. Controls were subjects enrolled in Center for Human Genome Variation studies through Duke Institutional Review Board approved protocols (n=830).
Identification of potentially causal variants
Sequence Variant Analyser (SVA)4 (http://www.svaproject.org/) was used to identify variants of interest using standard filtering criteria, (Single nucleotide variant (SNV) quality, SNV consensus score, insertion-deletion (INDEL) consensus score ≥20, INDEL quality ≥50, number of reads supporting SNV or INDEL ≥3). We designed screens to identify highly penetrant genotypes that might account for each child's conditions, and prioritised variants as follows: (1) homozygous (including hemizygous X variants) in the proband and never homozygous in the controls (recessive and X-linked variants); (2) heterozygous in the proband and absent in the parents and controls (putative de novo variants); and (3) from genes with two rare (MAF<0.03) variants in the proband that were not seen together in the parents or in any controls (compound heterozygotes). All variants, whether annotated as functional or not, were subjected to the screens for homozygous, X-linked and de novo candidates, the screen for compound heterozygous variants was limited to missense and nonsense SNVs, and frameshift INDELs. Appropriate functional work, where applicable, was performed based on the annotated function of the variant (online supplementary figure 1).
Further filtering of variants
Homozygotes
We removed any homozygous variant that was present in >3% of controls (corresponding to a disease frequency of 1 in 4500 or greater). For homozygous variants that were not present in the heterozygous form in both parents, we first removed those with low coverage (<10 reads), and then examined raw alignments for the remainder. In all cases, this was sufficient to resolve whether the variant was present in the parent but not called (because of <3 reads or poor quality scores), or incorrectly called as homozygous in the child.
De novos
Parental and proband raw alignments were examined for all potential de novo SNVs. The majority was ruled out for one of the following reasons: (a) low coverage in parents (<10x); (b) variant is visibly present in parental alignments but not identified by SAMtools; or (c) alignments look unconvincing (eg, multiple mismatches in same read, variant is at the very ends of reads) in proband and/or parents. For potential de novo INDELs, we removed those with fewer than five variant reads or with a variant/reference read ratio ≤0.3 (the vast majority) before inspection of raw alignments.
Compound heterozygotes
Raw alignments for all potential compound heterozygous variants were inspected in the proband and parents to ensure that the contributing variants were each inherited from a different parent.
Communication of results to families
All families underwent genetic counselling at the time of participation. In the initial counselling session, de novo, autosomal-recessive and X-linked inheritance patterns were discussed and it was emphasised that autosomal-dominant conditions with incomplete penetrance, synergistic heterozygosity, mitochondrial disorders and epigenetic changes would not be detected with this approach. All families were aware that a variant of interest that may be detected may not be definitely proven casual, and also that no results may be obtained. Parents were informed that variants of uncertain significance would not be reported to them. We debated if we should re-contact families after completion of the study, in the event that a variant of uncertain significance was subsequently thought to be casual but it was decided that it was not feasible to offer to do so. Variants thought to be causal or reasonably thought to contribute to the patient's phenotype were confirmed in a CLIA-certified laboratory prior to communication to the families, at which time a second genetic counselling session was arranged for discussion of results. With the permission of the families, the information was then communicated to the child's physicians.
For families wherein there would be no conclusive results, the second counselling session would be held after completion of the sequence data analyses. It was discussed with families that secondary or incidental findings in the child or the parents would not be intentionally screened for. If incidentally observed, the only variants that would be communicated were those within known genes that would result in premature death if untreated. Detection of carrier status in the affected child would not result in communication of such results. Detection of carrier status in the parents for known genetic conditions would be communicated to them, although it was emphasised that the genomes would not be proactively screened for such variants.
Results
Exome sequencing of each trio (table 1) resulted in an average coverage at captured regions of 71x (table 2). We used the SVA software,4 followed by manual inspection of candidate variants, as described in the online supplementary methods, to screen for candidate homozygous X-linked, compound heterozygous and de novo variants. The SVA screening produced a list of 260 candidate de novo SNVs and 364 candidate de novo INDELs, of which 18 SNVs (7%) and 2 INDELs (0.5%) were retained as high-confidence variants after manual inspection (table 2). Using this screening procedure, we found a likely genetic diagnosis in six of the families, a likely explanation for one of the clinical features in a seventh subject and a number of suggestive mutations in other families. No secondary (incidental) variants were detected in the probands or their parents.
Demographic and clinical features of sequenced patients
Exome sequencing quality and summary of rare homozygous and de novo variants
Likely genetic diagnosis: Trios 1 and 7—EFTUD2
Depending on how ‘functional’ mutations are defined, sequencing studies suggest an average of about one functional de novo mutation per genome.5 In this study, we see a total of 20 high-confidence de novo variants, somewhat higher than reported for controls.6 A particularly striking observation is that of these 20 de novo putatively functional variants, two were observed in the same gene, EFTUD2, in trio 1 and trio 7. Both variants were confirmed as de novo with Sanger sequencing. Very approximately, assuming (incorrectly) that each gene of the approximately 22 000 captured is equally likely to harbour a de novo mutation, the likelihood of seeing the same gene affected by chance in 2 of 20 de novos is 0.0086, suggesting the possibility of involvement of EFTUD2 in these patients' conditions. The patients share some clinical features (table 3), although they were not originally considered to be similar.
Clinical features of the two patients with EFTUD2 mutations, demonstrating similarities and dissimilarities between the two
The variant in trio 1 is a G/A transition located at the +5 position in the splice donor site of exon 11. G>A mutations of the +5G have been observed in several human inherited disorders,7–9 and in some studied examples site-directed mutagenesis of the +5G results in reduced splicing efficiency.10–12 Investigation of the mRNA isolated from blood of the proband and parents did not detect altered splicing or expression level, but tissue-specific impaired splicing remains a possibility. Documentation of a functional effect on splicing will be required to confirm pathogenicity of this variant. The EFTUD2 variant in trio 7 is a frameshift INDEL causing the premature termination of the protein at the end of exon 9 (residue 222/962). This study thus identified EFTUD2 as a leading candidate for explaining the conditions in these children. Subsequent to this work, Lines and colleagues13 very recently reported an analysis of 12 patients with Mandibulofacial Dysostosis with microcephaly, and found that all have de novo mutations in EFTUD2. On examination, both these patients show similarities to the children in this report, and the patient from trio seven fits the condition very closely.
Trio 2: NGLY1
Screening for compound heterozygous variants revealed that patient 2 had inherited a frameshift variant in the last exon of NGLY1 from his mother, and a nonsense mutation in exon 8 from his father. NGLY1 encodes N-glycanase 1, which is involved in the degradation of misfolded glycoproteins. N-glycanase 1 has not been associated with a specific disorder, but the phenotype of this child is consistent with a congenital disorder of glycosylation (table 1), and transferring isoelectric focusing and N-glycan analyses have been normal on repeated testing. To further explore the effect of these variants, we compared NGLY1 protein expression in leucocytes extracted from blood from the patient, his parents and three controls. Both parents showed reduced expression compared with controls, and the patient had barely discernible levels of NGLY1 (figure 1). Dysfunction of NGLY1 would be expected to result in abnormal accumulation of misfolded glycoproteins due to impaired degradation. In our patient, liver biopsy showed an amorphous unidentified substance throughout the cytoplasm, suggestive of stored material in the liver cells. It is to be noted that extensive testing for lysosomal storage had also been pursued in this child, and all the results had been normal. Further cellular assays are underway to better characterise this mutation.

Expression of endogenous NGLY1 protein in peripheral blood mononuclear cells from patient, parents and three unrelated healthy controls. The protein expression level in the patient is less than both parents and healthy controls. GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Trio 3: SMAD4
A de novo non-synonymous mutation was identified in SMAD4 in trio 3, resulting in an isoleucine to valine substitution at amino acid position 500 (I500V). This variant has recently been reported to be the causal variant in approximately half of all cases of Myhre syndrome, a clinically heterogeneous and rare developmental disorder. All other cases in these reports were caused by substitutions at the same position, including ile500thr and ile500met. Myrhe syndrome is characterised by variable short stature, short hands and feet, facial dysmorphisms, muscular hypertrophy, skin thickening, joint limitation, deafness and cognitive delay.14–16 Our patient did not present as a typical case. Although he has hearing loss, cognitive impairment and some of the characteristic facial dysmorphisms as well as ocular anomalies and congenital heart defects, he lacks some key diagnostic features including short stature, muscular hypertrophy, joint limitation, skin thickening and skeletal abnormalities. However, he is much younger than most reported patients, and it is possible that some manifestations such as joint stiffness, muscular hypertrophy and the skin thickening may emerge later. He also has scoliosis, which has not previously been described as a feature of Myhre syndrome. This case illustrates that with NGS, more early diagnoses and detection of patients with atypical presentations of Mendelian disorders would occur, resulting in widening of the phenotypic spectrum of these disorders.
Trio 5: TCF4
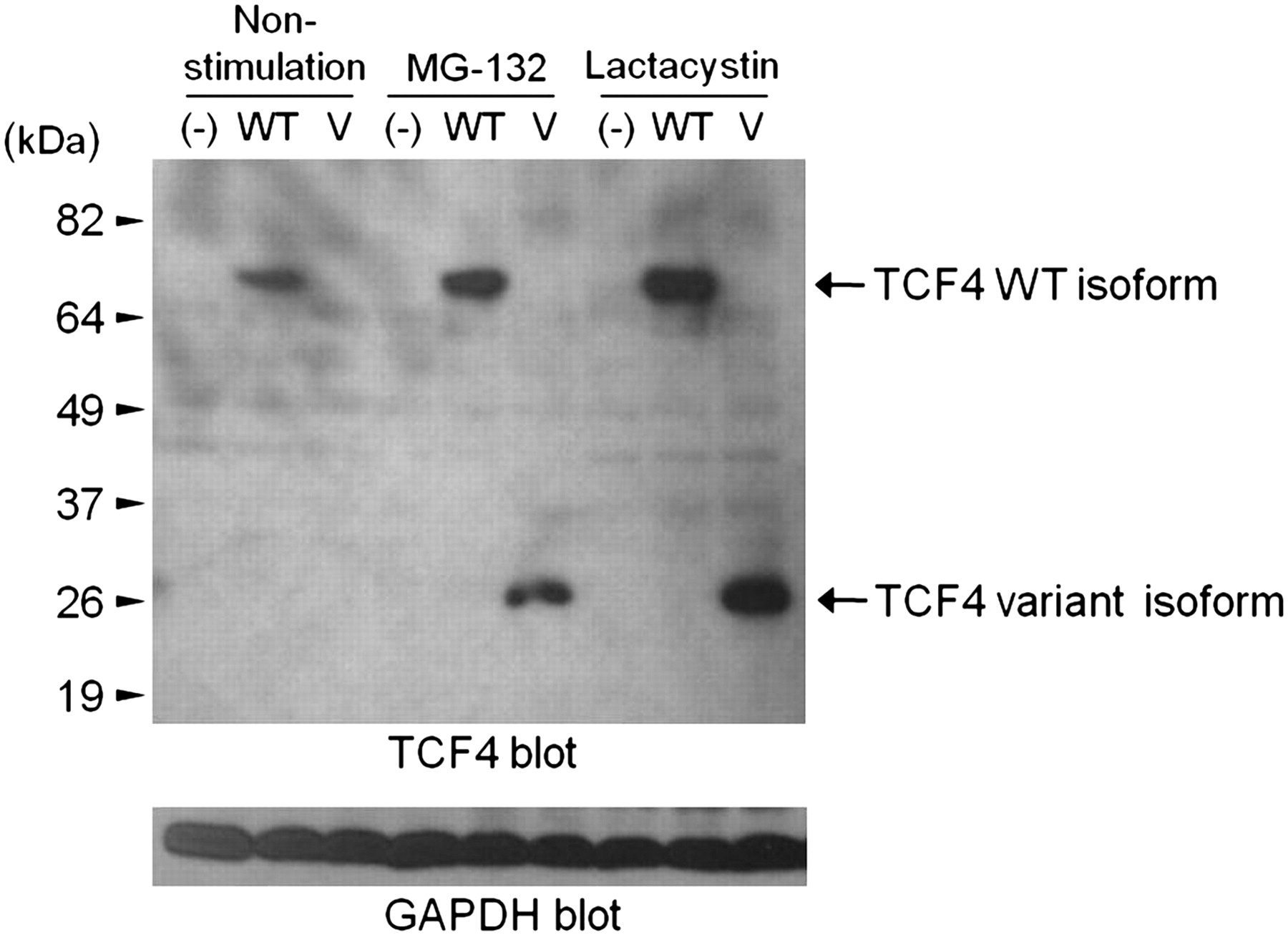
A novel de novo mutation was found in TCF4, a gene known to carry mutations responsible for Pitt-Hopkins syndrome (PHS). Sanger sequencing confirmed that the mutation is de novo, and a TaqMan assay in 1298 controls found no other carriers. We then evaluated the mRNA of the trio and found that the variant destroys the 3′ splice site of exon 9 (655 G>A, D219N), resulting in the incorporation of 37 incorrect amino acids before introduction of a stop codon and premature termination. Examination of protein expression showed that the variant protein was completely degraded through the ubiquitin-proteasome system (figure 2). This is likely to lead to haploinsufficiency of TCF4, the known cause of PHS.

Expression of TCF4 variant and wild-type (WT) protein in COS-7 cells. The variant protein (V) is only seen in the presence of proteasome inhibitors. GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
In retrospect, our patient's features of wide mouth, high cheekbones, deep-set eyes, limited speech and severe intellectual disabilities, are consistent with a diagnosis of PHS. She lacks the characteristic hyperventilation (seen in 86% of reported cases) and epilepsy (70%).17 Due to the absence of both these distinctive features, she had not been tested for this disorder, although it had been considered in the differential diagnosis.
Trio 11: SCN2A
A de novo variant was identified in SCN2A, a neuronal voltage-gated sodium channel gene. The mutation was at a site for which no previous mutation had been reported. Approximately 20 de novo and inherited variants in SCN2A have been reported to cause seizure disorders, mostly mild but occasionally accompanied by severe intellectual disabilities including infantile epilepsy.18–30 This non-synonymous SCN2A variant, Asp1598Gly, has a PolyPhen score of 0.99 (range 0–1), meaning that it is very likely to be detrimental to the protein,31 and was confirmed to be de novo by Sanger sequencing. Residue Asp1598 is located in transmembrane segment D4S3 of sodium channel Nav1.2, within the sequence WNIFDF that is highly conserved in mammalian and invertebrate voltage-gated sodium channels (figure 3). In the bacterial sodium channel, the corresponding sequence is WSLFDF, and the recently determined crystal structure indicates that this aspartate residue (D80) can form a hydrogen bond with a positive (arginine) gating charge in transmembrane segment S4.32 Conversion of this aspartate to the non-polar glycine residue would prevent this interaction, potentially impairing regulation of channel opening. These considerations strongly indicate the pathogenicity of this mutation.

{kind=link}
{kind=link}
{kind=link}
The SCN2A mutation, D1598G, is located in transmembrane segment 3 of the sodium channel protein domain 4. This residue is conserved in vertebrate, invertebrate DM (Drosophila) and bacterial (NaChBac) sodium channels. The D to Y mutation at the corresponding position of SCN1A was identified in a patient with severe myoclonic epilepsy (SME) of childhood, an early onset epileptic encephalopathy with features similar to the affected individual in trio 11 h, human; f, fish.
Further support for the role of this mutation comes from the closely related sodium channel SCN1A. SCN1A and SCN2A arose by gene duplication during vertebrate evolution, and retain 87% amino acid sequence identity (1747/2005) with most divergence in non-transmembrane domains. A de novo mutation in the corresponding residue of SCN1A, D1608Y, was found in a patient with severe myoclonic epilepsy of infancy, which like our patient is characterised by infantile seizures and intellectual disability.33 Three additional missense mutations in transmembrane segment D4S3 of SCN1A have been identified in patients with epilepsy (http://www.molgen.ua.ac.be/SCN1AMutations/), further demonstrating the pathogenic potential of this transmembrane segment of the protein.
SCN2A is not routinely included in DNA testing for epilepsy because mutations of SCN1A are much more common.
Interesting findings
In the remaining six cases, no variants judged as likely to be causal for most or all features were identified, although in two cases one or more interesting candidate variants were found.
Trio 4
Exome sequencing revealed several regions of homozygosity including several homozygous variants in EFEMP1 (two intronic SNVs and a 3′UTR INDEL), a gene in which heterozygous mutations are known to cause early onset maculopathies.34–36 Subsequent to this finding, it was judged that the patient's retinal phenotype of bilateral and symmetric distribution of drusenoid deposits most likely reflects dysregulation of the function of EFEMP1 (E Heon, personal communication). A real-time reverse transcriptase PCR assay indicated that the level of EFEMP1 expression in blood is too low to assess any effects of the variant on controls. This patient also carries a de novo non-synonymous coding SNV with a PolyPhen score of 0.999 in the gene ATP6AP2. This gene encodes the (pro)renin receptor and has multiple functions in the eye, heart, kidney, central nervous system and other tissues.37–39 This patient highlights the fact that some subjects who would undergo NGS may very well have more than one underlying diagnosis, and that all causative variants may not be detected.
Trio 6
A de novo variant was observed in the 5′ consensus splice site of exon 9 of the HNRNPU gene, which encodes HnRNP U. This gene is in the critical target region for the seizure phenotype of patients with microdeletion of 1q43–44,40 41 a highly variable syndrome characterised by speech delay, intellectual disability and seizures. In mice, HnRNP U has been shown to be linked to preaxial polydactyly caused by abnormal expression of SHH during limb development,42 and normal HnRNP U expression is essential for embryonic development.43 We have been unable to demonstrate a functional effect of the de novo variant in blood, but it remains possible that it affects expression of a particular isoform, perhaps in a tissue-specific manner during development. In addition, this patient has a de novo mutation in SMAD1, a gene that partners with SMAD4 in bone morphogenetic protein signal transduction.44 Given the association of de novo SMAD4 mutations with a spontaneous clinically heterogeneous developmental disorder (see above), it is possible that mutations in its close partner gene may cause similar phenotypes.
Interesting variants ultimately considered unlikely to be causal
Trio 2
A synonymous inherited X chromosome variant was found in the GPM6B gene, which has been considered a good candidate for causing cases of Pelizaeus-Merzbacher disease.45 Since Pelizaeus-Merzbacher Disease had been considered as a diagnosis for this patient, we tested the cDNA from the trio for possible effects on splicing, and the DNA from the maternal grandparents to examine the inheritance. We found that the mutation had no effect on the cDNA sequence of the patient, and that it was inherited from the paternal grandfather. This illustrates the importance of tracking candidate variants through the relevant pedigree before reaching a judgement concerning pathogenicity.
Trio 10
This patient has an inherited X chromosome variant predicted (by Genie46) to affect the 3′ splice site of exon 8 of ACSL4, a gene linked to intellectual disability with absent or severely delayed speech and dysmorphic facial features.47 However, cDNA sequencing revealed no differences between the patient and his parents. Combining this lack of function with a poor fit between the phenotype of the child and that associated with known mutations in the gene suggests that this variant unlikely to be responsible.
For the trios with no likely or suggestive causal variant, we will perform whole-genome sequencing to screen for variants that might have been missed by whole-exome sequencing such as exonic variants that were not captured, or structural variants not identifiable from exome data.
Discussion
This study highlights both the challenges and opportunities in the application of NGS to clinical diagnosis in patients with intellectual disabilities/congenital anomalies. In cases where we found a clear and likely cause of the condition, this conclusion depended on the knowledge of Mendelian diseases associated with the relevant genes. Two of these genes are already well known: TCF4 and SCN2A; however, the mutations we detected were novel. The example of EFTUD2 is of very particular interest. Before the recent identification of this gene, a possible case could be made on the basis of seeing de novo mutations in two of our patients, although we failed to show a functional effect of one of the two variants in the available tissue. Subsequent comparison of their phenotypes revealed a number of similarities. This example shows that a discovery paradigm focusing on a broad range of conditions provides an important complement to the more common current strategy of combining patients with similar conditions on strictly clinical criteria. By studying the genetics of a broader range of conditions as we did, it is possible to make a careful assessment of any phenotypic overlap of patients that have possible causal mutations in the same genes. In this way, it may be possible to identify conditions with broader phenotypic presentations than is possible in the strictly ‘phenotype first’ framework. However, we do note that confirmation that EFTUD2 is causal required its recent identification by Lines and colleagues.7 It is noteworthy that our study of only 12 patients pointed toward the possibility of EFTUD2 involvement in two of the cases. If a programme such as was used here were applied to many hundreds and eventually thousands of unexplained conditions, it is very plausible that many new genes would be nominated and confirmed using exactly this strategy.
Furthermore, information gained from genome sequencing as described here, focused on a broad range of patients, will likely expand the phenotypic spectrum of many currently well-known genetic disorders. Clinical decisions regarding whether or not to perform a genetic test largely depend on how well the patient fits the clinical description of the disorder. Although mutations in TCF4 are known to cause the well described PHS, the patient in this study did not exhibit two of the most common and differentiating symptoms of this disorder (periods of hyperventilation and seizures), and although the condition was considered, the diagnostic yield was not thought to be high enough to warrant testing. Similarly, the patient with the SMAD4 mutation that is known to cause Myhre syndrome did not show a typical manifestation of this syndrome. It is possible that there are many well described genetic conditions in which the variability in the phenotypic spectrum is not currently appreciated, and NGS may facilitate considerable broadening of this spectrum. The real power of diagnostic sequencing will depend on establishing very large databases that include mutations of interest and corresponding phenotypes. For example, intellectual disabilities and/or congenital abnormalities occur in approximately 3–4% of children,48 49 and a majority of these are due to underlying genetic causes, yet close to 50% of children with one or both of these phenotypes remain undiagnosed.50 51 It is likely that a high proportion of these undiagnosed cases will start to be sequenced annually in the next few years, creating the opportunity for very large databases that will permit the identification of currently unrecognised genotype-phenotype connections.
The suggestive finding for NGLY1 is also of particular interest. Rather than being a gene known to be responsible for a Mendelian disease with phenotypic similarity to the patient under study, this gene clearly acts in the same pathway as the known genes causing the Mendelian disorder that had been considered for the child, that is, a congenital disorder of glycosylation. This case illustrates how we can leverage known information about the function of a gene, and in particular its action within a pathway already implicated in Mendelian disease, to help identify new genetic diagnoses.
Our work also demonstrates the importance of the use of ‘general’, non-gene-specific functional evaluation of gene expression to confirm the pathogenicity of a variant. Since the de novo mutation in TCF4 had not been described before and involved a splice site, it made a strong but not definitive case for causality. Functional studies demonstrated that the mutation in TCF4 disrupts splicing and results in a protein targeted for degradation, which confirms causality. This work, therefore, helps to establish a general paradigm for such clinically motivated sequencing which includes not only the identification of candidate variants but also a generalised function evaluation of their impact on gene expression and splicing. However, as the number of sequenced patients increases, and as these data are increasingly shared in public databases, the need for functional work for some variants will decrease as the same variants are shown to occur in multiple patients with similar presentations (as for the SMAD4 variant in trio 3).
It is also important to emphasise that the paradigm we adopted in this study is likely to be similar to how NGS would be applied in clinical genetics practice, since general genetics clinics would have patients with widely differing phenotypes. Our study demonstrates the type of patients that would be sequenced in these clinics and provides data regarding expectations of finding a cause, the importance of functional assays for probable variants and the value of pre-screening patients to determine eligibility for NGS. With our inclusion and exclusion criteria, we set out to maximise the likelihood that an underlying undiagnosed genetic condition was present in each of the enrolled patients, and found causal or likely and interesting variants in 8/12 patients. It is also likely that in clinical practice, partial explanations would be detected for diverse manifestation in the same patient, as in trio 4 in our study, emphasising the complexity of genetic counselling for such a patient whose manifestations are likely to be due to more than one underlying genetic cause. Establishing a diagnosis is often of value even when a clear change in treatment is not indicated by the diagnosis. For example, close and ongoing observation for seizures is now indicated for patient 5 (TCF4), and avoidance of medications that may trigger seizures, such as antihistamines.52 The family can be informed that the disorder is due to a de novo variant, and in the absence of parental mosaicism, other family members are not at risk, and with future pregnancies the recurrence risk for the parents is low. Additionally, they can learn about PHS, have a better idea of future expectations, and reach out for support from families with similarly affected children. Similarly, patient 11 (SCN2A) should avoid common anti-epileptic drugs whose primary mechanism is sodium channel inhibition, since these exacerbate symptoms in patients with SCN1A mutations.53 A confirmed molecular diagnosis may also protect patients from incorrect diagnoses that could lead to unhelpful therapy options.
While cost benefit analyses were not the focus of this work, it is interesting to note that some of the patients who now have a genetic diagnosis, underwent many genetic tests prior to exome sequencing at a considerable estimated cost (eg, more than $22 000 were spent on laboratory investigations in Trio 2) While estimating the real costs of exome sequencing is difficult, it is already clear that in some cases, interrogating genes one by one or in panels will rapidly lead to greater total costs than exome or whole-genome sequencing. While these considerations are encouraging, as is the success rate of six likely genetic diagnoses out of 12 cases (with one further case likely explained partially), this work was performed in a research environment and there will be many challenges involved in a transition to fully clinically based applications. Itemising those challenges, from cost and reimbursement to the type and manner of communication to the families (including the issue of incidental findings), is beyond the scope of this work, but we would highlight two challenges in particular. First, in our experience, laboratory-based functional analysis is an important part of the evaluation, and it remains unclear how this would be incorporated into routine clinical application of NGS, even as NGS is beginning to be offered by commercial laboratories as a clinical test. Second, this work required substantial manual interrogation of both sequence data and candidate genes. Although variant calling procedures are continually improved and there are likely to be routines developed to simplify the process of candidate identification,54 it seems likely that for the foreseeable future, some level of expert judgement will continue to be required to identify causal mutations from sequence data, which will contribute to the cost and time of this type of diagnostics. Currently, it is difficult to imagine how the level of both variant inspection and functional evaluation could be provided as part of routine clinical diagnostic testing. These current essential functions, therefore, present a significant challenge to the use of NGS to provide genetic diagnoses.
Finally, we note that there are a number of reasons that causal variants may have been missed in some trios in this study. One important factor is that we do not have a comprehensive understanding of the function of most genes. For genes whose function is not well characterised, extensive functional follow-up may be required to assign causality to a de novo or homozygous variant carried by an individual patient. We may also fail to detect some causal variants. Exome sequencing does not capture all exons, nor non-coding regulatory regions, and structural genomic variants such as CNVs are difficult to recognise. Additionally, variants within captured regions may be missed by the mapping/variant-calling algorithms. In the future, we anticipate this approach will be improved by the use of whole-genome sequencing and improved variant identification, although for the foreseeable future a small proportion of the genome will remain refractory to high throughout sequencing. It is also possible that causal variant(s) may exert their effects through more complex inheritance patterns than investigated in this study.
In summary, this work indicates that the application of NGS should be strongly considered in all cases where a genetic condition is strongly suspected but traditional clinical genetic testing has proven negative. Furthermore, in some cases at least, it is likely that NGS will prove faster and less expensive than the long diagnostic odyssey many families now endure. However, our work, like that of others, offers the cautionary note that it will probably be possible to identify very strong candidate variants in any sequenced genome and that further studies such as functional assays or multiple patients with mutations in the same gene will often be needed to establish causality. Considerable attention must be paid to establishing appropriate standards of evidence before the results of NGS are used to influence patient care, and establishing such standards will be a major challenge for NGS in the clinic.
Acknowledgments
The authors would like to thank Min He for software development and support, Curtis Gumbs, Latasha Little and Ken Cronin for laboratory work and Janelle O'Brien for figure 3. Also, thanks to the following individuals and funding bodies for control samples: R Brown, K Welsh-Bomer, C Hulette, J Burke, E Pras, D Lancet, Farfel, E Ruzzo, K Pelak, R Radtke, A Husain, M Mikati, W Gallentine, S Sinha, D Attix, J M McEvoy, E Cirulli, V Dixon, N Walley, K Linney, E Heinzen, O Chiba-Falek, J P McEvoy, J Silver, M Silver, D Levy, H Meltzer, D Valle, J Hoover-Fong, N Sobriera, C Manzini, A Poduri, N Calakos, C Depondt, S Sisodiya, G Cavalleri, N Delanty, P Lugar, W Lowe, S Palmer, D Marchuk, D Daskalakis, M Winn, A Holden, E Behr, S Kerns, H Oster, R Murdock, The Murdock Study Community Registry and Biorepository Pro00011196, J Milner, Ellison funding, ARRA 1RC2NS070342-01, Bryan ADRC NIA P30 AG028377, NIH Research Grant NS34509, NIMH Grant RC2MH089915, Division of Intramural Research, NIAID, NIH. Most importantly we would like to thank all patients and their families for their participation in this research.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
AN and VS contributed equally to this work.
Competing interests None.
Patient consent Obtained.
Ethics approval Ethics approval was granted by the Duke University Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Inquiries from scientists/clinicians about specific variants, variants in specific genes, or putatively pathogenic groups of variants (eg, rare recessive) should be addressed to the corresponding author and information will be provided in compliance with the signed consents of the participating families.