Article Text

Abstract
Background NLRP7 mutations are responsible for recurrent molar pregnancies and associated reproductive wastage. To investigate the role of NLRP7 in sporadic moles and other forms of reproductive wastage, the authors sequenced this gene in a cohort of 135 patients with at least one hydatidiform mole or three spontaneous abortions; 115 of these were new patients.
Methods/Results All mutations were reviewed and their number, nature and locations correlated with the reproductive outcomes of the patients and histopathology of their products of conception. The presence of NLRP7 mutations was demonstrated in two patients with recurrent spontaneous abortions, and some rare non-synonymous variants (NSVs), present in the general population, were found to be associated with recurrent reproductive wastage. These rare NSVs were shown to be associated with lower secretion of interleukin 1β and tumour necrosis factor and therefore to have functional consequences similar to those seen in cells from patients with NLRP7 mutations. The authors also attempted to elucidate the cause of stillbirths observed in 13% of the patients with NLRP7 mutations by examining available placentas of the stillborn babies and live births from patients with mutations or rare NSVs. A number of severe to mild placental abnormalities were found, all of which are known risk factors for perinatal morbidity.
Conclusions The authors recommend close follow-up of patients with NLRP7 mutations and rare NSVs to prevent the death of the rare or reduced number of babies that reach term.
- Hydatidiform mole
- NLRP7
- reproductive wastage
- spontaneous abortion
- stillbirth
- genetics
- molecular genetics
- immunology (including allergy)
- obstetrics and gynaecology
- reproductive medicine
Statistics from Altmetric.com
- Hydatidiform mole
- NLRP7
- reproductive wastage
- spontaneous abortion
- stillbirth
- genetics
- molecular genetics
- immunology (including allergy)
- obstetrics and gynaecology
- reproductive medicine
Introduction
Hydatidiform mole (HM) is an aberrant human pregnancy characterised by abnormal embryonic development, hydropic degeneration of chorionic villi, and proliferation of the trophoblast. The common form of this condition occurs once in every 250 pregnancies in Western societies.1 Common moles are sporadic, usually not recurrent, and have a complex aetiology involving both genetic and environmental factors. Among women with one mole, 1–6% will develop a second mole,2–7 depending on populations and studies, and 10–25% will experience a second reproductive wastage, mostly as a spontaneous abortion (SA).2 4 8 9
Molar pregnancies are first diagnosed on the basis of ultrasonography and high serum concentrations of β-human chorionic gonadotropin. Definitive diagnosis is made after histopathological examination of the evacuated products of conception (POCs), which allows them to be divided into complete and partial moles and distinguished from non-molar SAs. At the histopathological level, complete HMs (CHMs) do not contain embryonic tissues other than the chorionic villi and have excessive trophoblast proliferation.10 Partial HMs (PHMs) may contain other embryonic tissues (amnion, chorion or other) but have mild and focal trophoblastic proliferation.10 Non-molar SAs may contain embryonic tissues, but most do not have trophoblastic proliferation. Because histopathology is a qualitative science and lacks quantitative measurements to assess the degree and extent of trophoblastic proliferation (mild, excessive, focal, occasional, etc), there is wide interobserver and intraobserver variability, mainly in distinguishing PHMs from SAs and less often in distinguishing CHMs from PHMs.11 In addition, epidemiological studies have shown that the frequency of moles is higher in patients with recurrent SAs than in women from the general population.12 13 Also, a history of recurrent SAs is a well-known risk factor for moles.14 Furthermore, women with recurrent moles may have CHMs, PHMs and SAs, indicating that these three histopathological entities have, at least in some cases, the same underlying aetiology and are a continuous spectrum of the same condition.
To date, one gene, NLRP7, has been shown to have a causal role in recurrent HMs and associated reproductive wastage.15 Mutations in this gene have been reported by various groups and in patients from several populations, demonstrating that NLRP7 is a major gene for this condition16–22 (http://fmf.igh.cnrs.fr/ISSAID/infevers/). Patients with familial recurrent HMs have two defective alleles and can be homozygous for the same mutation or compound heterozygous for two different mutations. To date, only seven patients with a single defective allele in NLRP7 have been reported.16 17 NLRP7 (NOD-like receptor proteins, pyrin containing domain 7) is formed from three main domains: a pyrin domain, a NACHT domain (found in the NAIP, CIITA, HET-E and TP1 family proteins), and 9–10 leucine-rich repeats (LRRs) depending on splice isoforms. NLRPs are a subfamily of proteins with roles in inflammation and apoptosis.23 In vitro, NLRP7 overexpression inhibits caspase-1-dependent interleukin 1β (IL1B) secretion.24 Recently, we demonstrated that peripheral blood mononuclear cells (PBMCs) from five patients with different NLRP7 mutations secrete lower levels of IL1B and tumour necrosis factor (TNF) than PBMCs from controls (Messaed et al, unpublished work). We showed that, despite their low cytokine secretion, the intracellular levels of mature IL1B in PBMCs of one analysed patient was normal, comparable to that of control cells, indicating a defect after IL1B processing, most likely affecting its secretion into the extracellular milieu. We suggested that the impaired cytokine secretion and consequent inflammatory response of the patients, due to their NLRP7 mutations, make them tolerant to the growth of the aberrant molar conceptions with no embryo, and the delayed rejection of these conceptions contribute to the molar phenotype.
Because of the diagnostic overlap between moles and SAs, we widened our selection criteria for NLRP7 sequencing and included patients with at least one HM (≥1 HM) or three SAs (≥3 SAs). Here we report NLRP7 mutation analysis in 135 unrelated patients with a spectrum of reproductive wastage. We show that the highest frequency of NLRP7 mutations is found in patients with ≥2 HMs and the lowest in patients with ≥3 SAs. We demonstrate a significant association between complete moles and the presence of at least one protein-truncating mutation. We show that rare non-synonymous variants (NSVs) in NLRP7 confer genetic susceptibility for recurrent reproductive wastage. Finally, we show that patients with NLRP7 mutations and rare NSVs have variable degrees of placental abnormalities associated with increased perinatal morbidity.
Patients and methods
Patients and controls
The study was approved by the institutional review boards of McGill and collaborating institutions. All patients and controls provided written consent to participate in this study. Patients were ascertained by (i) referral to us from various collaborators for genetic testing, (ii) recruitment from the miscarriage clinic of the McGill University Health Centre, and (iii) referral from the Quebec and Montpellier registries of trophoblastic diseases. Selection criteria were either at least one HM (≥ 1 HM) or one trophoblastic disease or the occurrence of at least three SAs (≥3 SAs). For most patients, clinical information was collected using standard pro forma recapitulating complete reproductive, medical and family histories. One patient, 428, with one NLRP7 mutation previously reported by our group16 had a prematurely born baby at 28 weeks in her last pregnancy. The baby was later diagnosed to have several congenital abnormalities including bilateral clubfoot, intraventricular haemorrhage grade II on the left side of the brain, developmental delay, mild tracheomalacia and patent ductus atresia which required several operations. Blood karyotype analysis, at a resolution level of 400 bands, revealed a 46,XY normal karyotype in 11 analysed metaphases.
For mutation analysis, control DNA was from women either from the Centre d'Etude du Polymorphisme Humain (CEPH) families or families of European descent with various inherited conditions, unrelated to pregnancy losses, and with 5–16 children. Beside women from the CEPH families, most controls were of Canadian origin, with a significant number being of French ancestry. However, their complete reproductive history and whether they had had reproductive wastage is not known.
Mutation analysis and annotation
Mutation analysis was performed, as previously described,20 by PCR amplification of genomic DNA of the 11 NLRP7 exons followed by direct sequencing in the two directions. Sequences were analysed using DNASTAR. In the text, we use the term mutations to indicate DNA changes, leading to protein truncations or non-synonymous changes that were not found in any of the tested controls including those of the same, or related, ethnicities to the patients. We use the term non-synonymous variants (NSVs) to indicate coding DNA changes leading to amino acid changes that were found in controls from any ethnic group. When possible, identified mutations and variants were phased by genotyping available parents and other family members. Nucleotide numbering for mutations and variants uses cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence, NM_001127255.1.
Cytokine secretion and western blotting
PBMCs were separated using Ficoll, counted, and cultured in the absence or presence of ultrapure lipopolysaccharides (1 μg/ml; Cedarlane 423(LB) from Escherichia coli 0552:B5) for 24 h. Supernatants were collected and assayed by ELISA for IL1B and TNF secretion (BD Biosciences, Becton Dickinson Canada Inc, ON, Canada). Statistical analyses were performed using analysis of variance single-factor analysis, as it is the appropriate comparison test to deal with a single independent variable between all the compared values. p<0.05 was considered to be significant. Western blot analysis was performed using monoclonal antibodies directed against human IL1B (Cell Signaling Technology, USA), α-tubulin (Cell Signaling Technology, New England Biolabs, ON, Canada) (1:1000 dilution) and β-actin (Chemicon International, Millipore, Massachusetts, USA). Protein bands were revealed using Hyperfilm ECL Western blotting detection reagents (GE Healthcare, Mississauga, ON, Canada) and quantified by Image J software (http://rsb.info.nih.gov/ij/).
Histopathology
For histopathological diagnoses of CHM, PHM and SA, 105 tissue sections from 31 POCs were stained with H&E, examined independently by two pathologists with expertise in early pregnancies, scored for four parameters (presence of nucleated red blood cells inside chorionic villi, presence of fetal membranes or tissues beside chorionic villi, degree of trophoblast proliferation, and degree of hydropic change), and classified as CHM, PHM or SA. For late and term placentas, tissues were screened independently by two other pathologists with expertise in term placentas.
Results
NLRP7 mutations in the spectrum of reproductive wastage
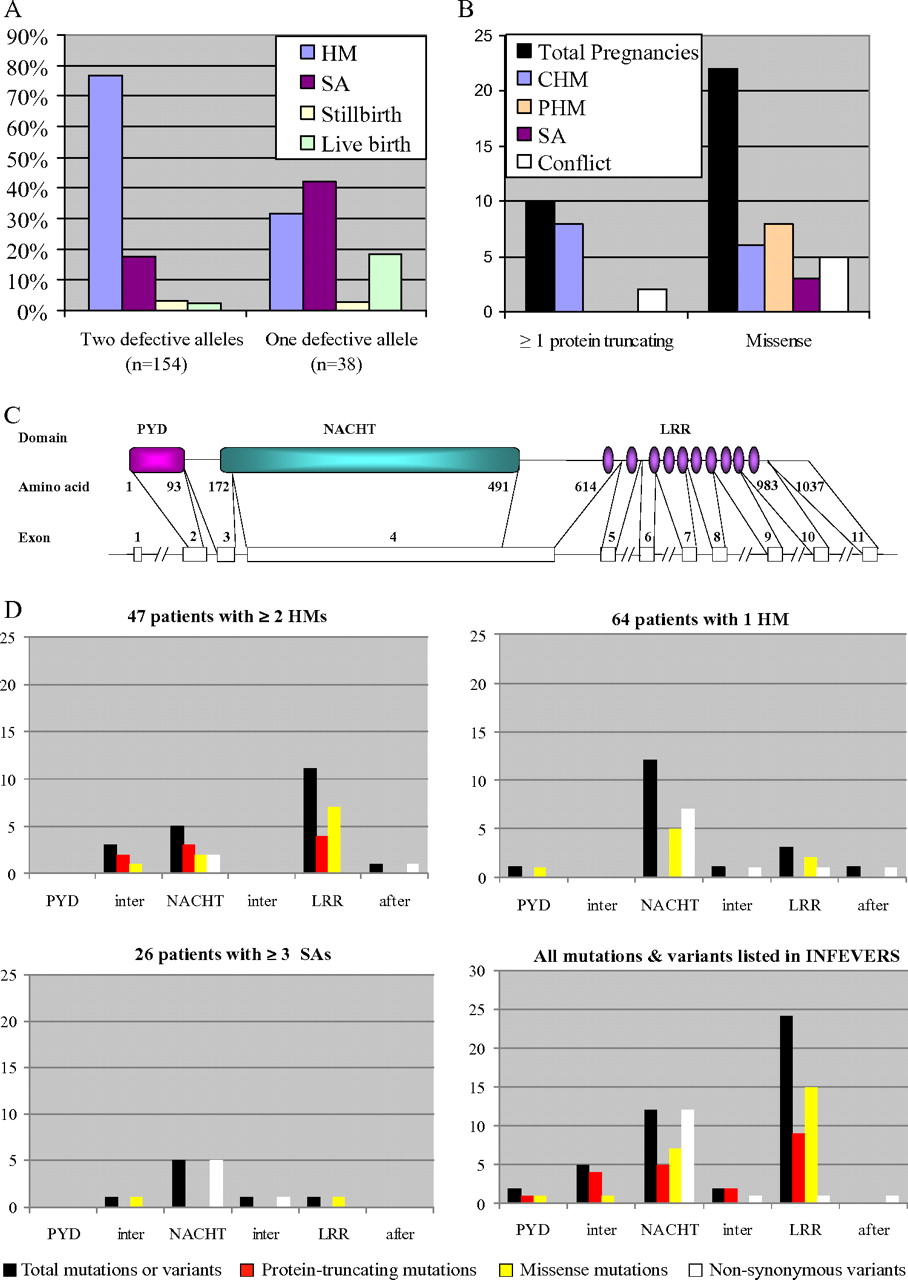
In this study, we sequenced NLRP7 in 135 unrelated patients with ≥1 HM or ≥3 SAs; 115 of these were new patients. Of the 135 unrelated patients, 45 had had ≥ 2 HMs, 64 had had only one HM (with or without other reproductive wastage), and 26 had had ≥3 SAs (figure 1). The highest frequency of mutations was found in patients with ≥2 HMs (60%; 26/45 patients), followed by patients with one HM (13%; 8/64 patients), then patients with ≥3 SAs (8%; 2/26 patients). Of the eight patients with one HM and NLRP7 mutations, six (75%) had had at least two additional instances of reproductive wastage, one had had one mole and two live births, and the remaining had had one mole, but no data are available about her other reproductive outcomes. This indicates that NLRP7 mutations predispose patients to recurrent reproductive wastage rather than sporadic moles. Of the 26 women with ≥3 SAs, two had NLRP7 mutations. One had had two live births and seven SAs and was heterozygous for R156Q. The second patient had had four SAs, one of which led to a gestational trophoblastic disease that required methotrexate treatment, and was heterozygous for A719V. Both mutations, R156Q and A719V, were previously seen in women with moles16 and were not found, respectively, on 300 and 200 chromosomes from control women of matching ethnicity to that of the two patients (both of European descent). This demonstrates that NLRP7 is also responsible for some cases of recurrent SAs and that the variability in the reproductive outcomes of patients with the same mutation may be due to the genetic background of the patients, environmental factors, or both. Our data are in agreement with previous epidemiological data showing the occurrence of a second case of reproductive wastage in 10–25% of women with a prior HM.2 4 8 9

Summary of NLRP7 mutation analysis of 135 unrelated patients with a spectrum of reproductive wastage. HM, hydatidiform mole; NSV, non-synonymous variant; SA, spontaneous abortion.
Eight new mutations and variants including three protein-truncating ones
Of 135 unrelated patients sequenced, 36 had at least one protein-truncating mutation or NSV that were not seen in controls of matching ethnicity. Among these mutations/variants, there were 28 different mutations/variants, 20 of which have previously been reported,15–22 25 and eight were new mutations or variants. Three of the new mutations were protein-truncating (c.930_931del, p.Gln310HisfsX38; c.1018_1020delinsCAAAA, p.Glu340GlnfsX11; c.2791_2792delTG, p.Cys931X) and most likely pathogenic. The remaining five were missense (c.750C→A, p.Phe250Leu; c.1018G→A, p.Glu340Lys; c.1169G→A, p.Arg390His; c.1237C→T, p.Arg413Trp; c.2444G→A, p.Arg815His), and none was found in controls from several ethnic groups (online supplementary table 1). These five missense variants could (i) be pathogenic, (ii) increase the susceptibility of the patients to reproductive wastage, or (iii) be simply very rare variants that were not seen in our cohort of controls from the general population. Future studies are needed to clarify their status with respect to reproductive wastage. We note that none of these new missense variants is reported on the 1000 Genome Database website. Using Polyphen 2 software that predicts possible effects of an amino acid substitution on the structure and function of a protein using structural and comparative considerations, two missense mutations, Glu340Lys and Arg413Trp, were predicted to be probably damaging, while the remaining three, Phe250Leu, Arg390His and Arg815His, were predicted to be benign (table 1). The reproductive outcomes of the patients with the new mutations and variants are shown in online supplementary table 2.
Correlation between predicted effects of missense mutations and variants and reproductive outcomes of the patients
Genotype–phenotype relationships of mutations in NLRP7
Patients with one NLRP7 defective allele have better reproductive outcomes
Of the 36 unrelated patients with mutations from the three groups (≥ 2 HMs, 1 HM or ≥3 SAs), 24 had two defective alleles and 12 had a single defective allele. Comparing the reproductive outcomes of patients with one or two defective alleles (including related patients in familial cases) revealed that patients with one defective allele had significantly more live births (18.4% vs 2.5%), more SAs (37% vs 17.4) and fewer HMs (34.1% vs 73.5% of their pregnancies) than patients with two defective alleles (Fisher exact test, p value=2.809e–06) (figure 2A). Of the 12 patients, each with one defective allele, seven had had a single mole with other forms of reproductive wastage. These data indicate that patients with one identified defective allele most likely have a single defective allele and consequently better reproductive outcomes than those with two mutated alleles, who have a much greater chance of having recurrent molar pregnancies.

(A) Comparison of reproductive outcomes between patients with one or two defective alleles. n indicates the total number of pregnancies of patients from each category. In this figure, only major categories of reproductive wastage are shown; the other rare forms (blighted ovum, ectopic pregnancy, elective abortion, malformed baby) observed in small numbers (between 2 and 6) were either removed or merged with related categories. (B) Correlation between the nature of NLRP7 mutations and the histopathological diagnosis of products of conception (POCs) established by two pathologists. (C) Genomic and protein structures of NLRP7. (D) Distribution of the different types of NLRP7 mutations in its three domains. Distribution of mutations and variants found in our three categories of patients and of mutations listed on INFEVERS (http://fmf.igh.cnrs.fr/ISSAID/infevers/). After, indicates amino acids after the LRR domain; CHM, complete hydatidiform mole; Conflict, indicates two different diagnoses by the two pathologists; HM, hydatidiform mole; Inter, indicates amino acid between two domains; LRR, leucine-rich repeat; NACHT, found in the NAIP, CIITA, HET-E and TP1 family proteins; PHM, partial hydatidiform mole; PYD, stands for the Pyrin domain; SA, spontaneous abortion.
Protein-truncating mutations are associated with repeat CHMs
To investigate possible correlations between the nature of the mutations and the histopathological types of the molar tissues, 105 tissue sections from 31 POCs from 13 patients (from 12 unrelated families) with NLRP7 mutations were examined, scored and diagnosed independently by two pathologists. Both pathologists noted that recurrent molar tissues from patients with NLRP7 mutations have, in general, less trophoblastic proliferation than common sporadic moles. There was an agreement between the diagnoses of the two pathologists in 80% of the cases. Of 10 HMs from patients with at least one protein-truncating mutation, nine were diagnosed as CHMs by the two pathologists (figure 2B). Patients with missense mutations had more variability in the histopathological diagnosis of their POCs (figure 2B). These data show that repeat CHMs is the most severe phenotype caused by NLRP7.
The association between CHMs and protein-truncating mutations prompted us to compare the frequencies of protein-truncating mutations in all reported familial and singleton cases of recurrent moles with the hypothesis that, if protein-truncating mutations were associated with the severe phenotype, we would expect them to be higher in familial than singleton cases, since their presence would have favoured the manifestation of moles in all family members carrying them. A recapitulation of these cases showed a higher frequency of protein-truncating mutations in familial than singleton cases (52.17% vs 32.71%; online supplementary table 3). Although this association is not statistically significant, it indicates that some missense mutations are less penetrating and consequently not all family members carrying them manifest moles.
Distribution of mutations and variants in the three NLRP7 domains
The distribution of the different mutations and variants found in the three NLRP7 domains is shown in figure 2C, D. In this cohort of patients, protein-truncating mutations were only found in patients with at least two HMs. Patients with only one HM or at least three SAs had only missense variants (figure 2D). The distribution of all NLRP7 mutations and variants in our patients is similar to those reported by all groups (including ours), which are listed on INFEVERS (http://fmf.igh.cnrs.fr/ISSAID/infevers/) (figure 2D). The highest number of missense mutations (62%) is observed in the LRRs, which represent only 36% of the total size of the protein. These data confirm the importance of the LRRs for the normal function of NLRP7, as previously suggested.22 This suggestion is further corroborated by the fact that only one NSV, V699I, in the LRRs was seen among all controls we analysed from several ethnic groups compared with 12 in the NACHT domain (319 amino acids), which is even 15% shorter than the LRR domain (369 amino acids) (figure 2D).
The functional consequences of all missense mutations predicted by Polyphen 2 (http://genetics.bwh.harvard.edu/pph/) showed the association of missense variants with mild predicted functional consequences with the category of one HM or ≥3 SAs, while severe mutations were associated with the occurrence of ≥2 HMs (table 1).
Rare NSVs in NLRP7 are associated with recurrent HMs and SAs
To investigate whether the non-synonymous NLRP7 variants predispose women to reproductive wastage, we removed from our cohort of 135 women those with at least one mutation and those of non-European origin and compared the frequencies of NSVs in 53 European patients with no mutations in NLRP7 and 155 controls of European descent (table 2). This analysis revealed a statistically significant association between c.1441G→A, p.A481T and reproductive wastage (χ2=6.24, p=0.012). Of the 53 patients, 12 had had a single HM with either no data about their other reproductive outcomes or with normal pregnancies. When these cases were removed from the sample and only cases with one mole and at least another case of reproductive wastage or patients with ≥3SAs were included, the association with A481T was more significant (χ2=7.435, p=0.0063). Five other rare NSVs were more common in the patients than the controls, but did not individually reach statistical significance (table 2). We then looked for association between the presence of any of the NSVs or any of the rare NSVs, listed in table 2, and reproductive wastage and found significant association with patients with one HM or ≥ 3SAs (p=0.0003), which was even higher after removal of cases with one HM and no other reproductive wastage (p=0.000012). We note that none of the patients with two NLRP7-defective alleles had any of the rare NSVs. Among patients with a single defective allele, three had one or several rare NSVs. Parental DNA was available in one case, which allowed us to show that the rare variant is inherited on a different parental haplotype from the mutation. Taken together, our data support the role of A481T and the other rare NSVs in the genetic susceptibility for recurrent reproductive wastage.
Frequencies of non-synonymous NLRP7 variants in patients and controls of European descent
Low IL1B and TNF secretion by mononuclear blood cells from patients with A481T
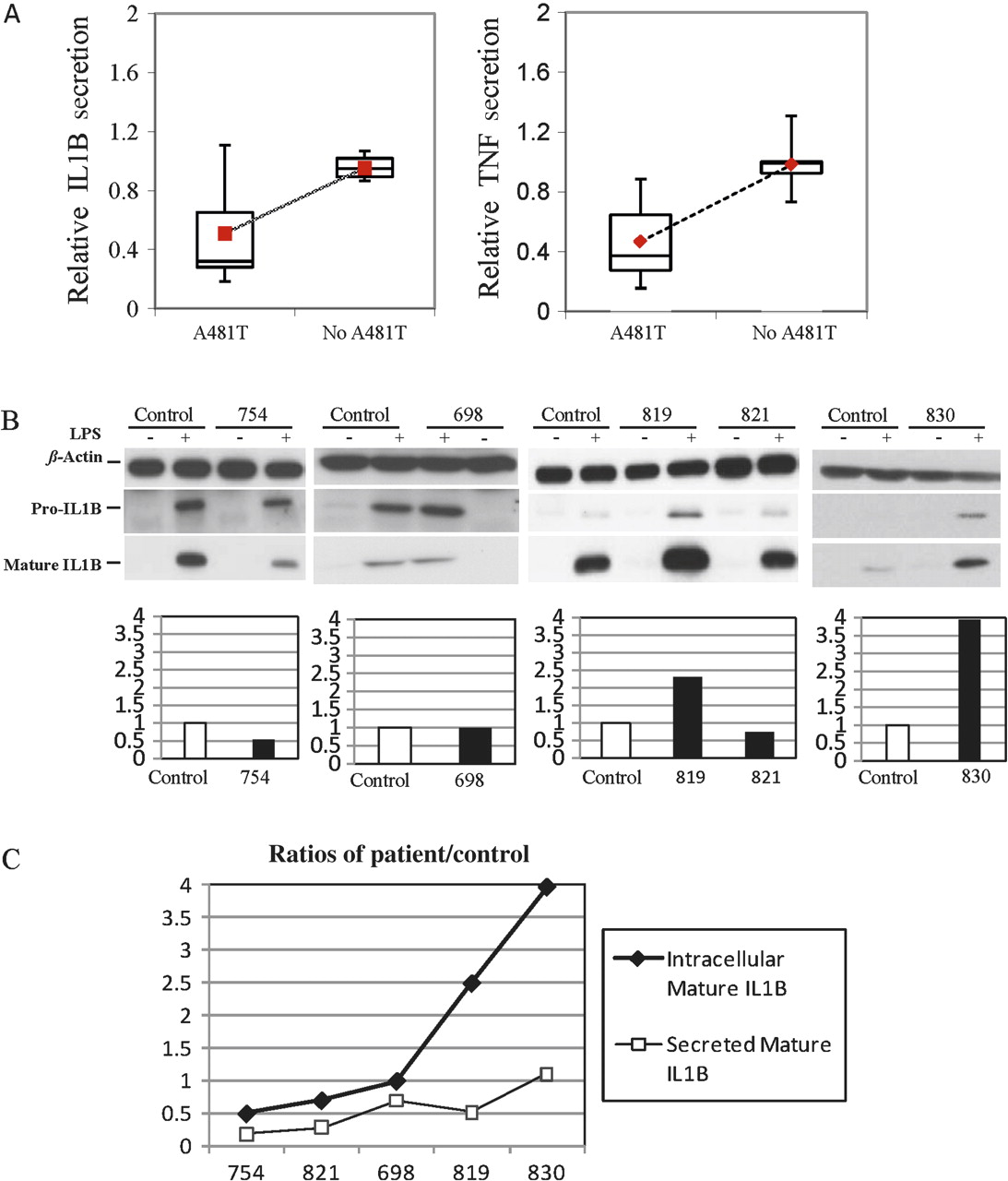
Recently, we have shown that PBMCs from five patients with NLRP7 mutations have reduced levels of IL1B and TNF secretion despite the fact that they have normal concentrations of intracellular mature IL1B (Messaed et al, unpublished work). To investigate the potential functional consequences of A481T and the other rare NSVs on IL1B and TNF secretion, PBMCs from five patients (698 (p.[V319I;A481T;G487E];[=]), 754 (p.[A481T];[=]), 819 (p.[A481T];[=]), 821 (p.[Q310R(;)L311I(;)A481T]), 830 (p.[A481T];[=])) carrying A481T with and without other NSVs and seven different controls without A481T and any of the other rare NSVs were stimulated ex vivo, and cytokine concentrations were determined by ELISA. This analysis showed that patients' cells secrete significantly lower levels of IL1B and TNF than control cells (p<0.001) (figure 3A). We then looked at the levels of intracellular production of pro and mature IL1B by cells from these five patients and one control using western blot analysis. We found variable concentrations of intracellular pro and mature IL1B in different patients (figure 3B) similar to those reported in healthy subjects.26 In all the analysed patients, the intracellular concentrations of mature IL1B mirrored those of pro-IL1B, demonstrating that NSVs in NLRP7 do not impair IL1B cleavage. This result is in agreement with our previous observation in one patient with a mutation, G380R, in NLRP7 (Messaed et al, unpublished work). We then looked at the ratios of intracellular and extracellular IL1B between cells from each of the five patients and the same control cultured, stimulated and assayed at the same time. We found that the ratios of secreted IL1B by patient cells relative to control cells (patient/control) were lower than the ratios of their intracellular mature ILIB (figure 3C). These findings are in agreement with our previous observation in a patient with a mutation in NLRP7 and demonstrate that A481T and the other rare NSVs have functional consequences and reduce cytokine secretion upon stimulation with lipopolysaccharides. Taken together, these data show the association of A481T and rare NSVs with lower cytokine secretion similar to those observed in patients with NLRP7 mutations, and support further the role of these rare NSVs in conferring genetic susceptibility for reproductive wastage.
{kind=link}
{kind=link}
{kind=link}
(A) Interleukin 1β (IL1B) and tumour necrosis factor (TNF) secretion by ex vivo stimulated peripheral blood mononuclear cells (PBMCs) with lipopolysaccharides (LPS) from five patients with A481T and other rare non-synonymous variants (NSVs) in NLRP7. The patients included in this analysis were 698, 754, 819, 821 and 830. Each patient had one copy of A481T with or without other rare NSVs (online supplementary table 4 and supplementary figure 2). PBMCs from seven subjects without A481T and any of the other rare NSVs were used as controls. Patients carrying A481T and other rare NSVs secrete significantly lower amounts of IL1B and TNF than controls not carrying these variants (p<0.0001). (B) Cell lysates of LPS-stimulated PBMCs from five patients and the same control were subjected to immunoblotting to determine the concentrations of pro-IL1B and mature IL1B. The concentrations of mature IL1B were normalised to those of β-actin. IL1B is not constitutively expressed by PBMCs. Upon stimulation, PBMCs from the patients and control produced variable amounts of intracellular pro and mature IL1B as reported in healthy subjects, and none had defective IL1B processing. (C) Comparisons were performed between the patient/control ratios of intracellular mature IL1B, quantified by the Image J software, and the patient/control ratios of secreted IL1B in the extracellular milieu measured by ELISA. This analysis shows that patients' cells secrete lower amounts of the produced mature intracellular IL1B than those of controls.
Increased perinatal morbidity and placental abnormalities in patients with NLRP7 mutations or rare NSVs
To date, six of our 46 patients with at least one NLRP7 mutation (13% of our patients) had had seven stillbirths (3.4% of their pregnancies). Tissues from all the placentas of these stillborn babies were not available to us for evaluation. The descriptions provided by the patients for four cases indicated the death of morphologically normal babies (online supplementary table 5). Medical reports were available for two cases (online supplementary table 6). In one case, the patient manifested, at 26 weeks of gestation, pre-eclampsia and placental abruption. Infarction and calcification were diagnosed by histophathological examination of the placenta of the delivered baby, who died later. In the second case, a placental haematoma was diagnosed by ultrasonography after the intrauterine demise of the baby.
In an attempt to understand what caused the death of these babies, we retrieved placental tissues from one stillbirth and eight live births from six patients with NLRP7 mutations or rare NSVs, all of whom were living in Canada. Histopathological evaluation of the placentas by two pathologists with extensive expertise in term placentas revealed a number of abnormalities (table 3). This evaluation showed that only two of the nine analysed placentas did not have any abnormality, and six had one to several abnormalities of variable severity. These abnormalities included: mild to severe chorioamnionitis, inflammation of the chorion and amnion, which was seen in three placentas (online supplementary figure 1); mild chorangiosis, a placental sign associated with prolonged hypoxia, which was seen in three placentas; decidual necrosis, which was seen in three placentas; and dysmature stem chorionic villi, seen in two placentas (online supplementary figure 1). The abnormalities seem to correlate with the severity of the allele, with severe abnormalities in patient 428, who had a mutation and milder abnormalities than patients with rare NSVs.
Summary of histopathological evaluations of the placentas of patients with NLRP7 mutations and at least one rare non-synonymous variant
We have previously shown that patients with NLRP7 mutations and variants have postzygotic cleavage abnormalities. To investigate whether the abnormalities observed in the placentas from the stillborn and malformed male babies of patient 428, with a mutation in NLRP7, were caused by placental mosaicism and the presence of androgenetic cells with two X chromosomes, we used fluorescent in situ hybridisation and probes for the Y and X chromosomes. This analysis did not reveal any XX cells in five available sections from the two placentas, and all cells from the two placentas had normal numbers of sex chromosomes with one X and one Y. We then searched for other chromosomal aneuploidies with probes from four autosomes and did not identify mosaicism or aneuploidies in the available tissues from the two placentas of patient 428.
Discussion
In this study, we report NLRP7 mutation analysis in a cohort of 135 women with at least one HM or three SAs, of which 115 were new patients. We show genotype–phenotype correlations between the number of defective alleles, their nature and their locations with the reproductive outcomes of the patients. We found that two defective alleles were associated with more severe reproductive outcomes than one defective allele. Also, protein-truncating mutations were associated with CHM and were more common in familial than singleton cases of recurrent moles. Because of their severity, protein-truncating mutations are less likely to be modulated by the genetic background of the patients or environmental factors and consequently manifest as CHM in all family members carrying two protein-truncating alleles. In addition, the relatively more important trophoblastic proliferation in patients with protein-truncating mutations will favour the histopathological diagnoses of the POCs as complete moles by pathologists and consequently favour their familial manifestation. However, missense mutations were, in general, less severe and associated with more variability in the reproductive outcomes of the patients. For instance, mutations R156Q and A719V, found here in two patients with several SAs, were previously reported in two patients with moles, 647 and 636.16 Of the two previously reported patients with moles, one had had one live birth and three moles, two of which were PHMs, and the second patient had had two live births, one partial mole and one SA, and other earlier forms of fetal loss.16 Moreover, variants Q310R, L311I and A481T were found in one patient with one CHM and one ectopic pregnancy, while her mother and sister carrying the same variants on the same haplotype had had only SAs (case MoSw177, online supplementary figure 2). Taken together, these data confirm the overlap between the aetiologies of SAs and moles and indicate that other genetic and environmental factors modulate the defect.
Of the patients analysed, 41.9% did not have any mutation, but had one or several NSVs. Comparing the frequencies of each of the NSVs, individually or combined, in our cohorts of European patients and controls showed a significantly higher association in the patients. These associations were stronger when patients with sporadic moles and no other reproductive wastage were removed. Despite the fact that we included in this analysis all the patients referred to our laboratories who fitted our criteria (≥1 HM or ≥3 SAs), two factors may have biased the ascertainment of our samples: (i) our historical interest in the pathology of recurrent moles and (ii) the excellence of McGill Reproductive Centre in reproductive medicine. These two factors may both have favoured the ascertainment of severe cases with an underlying genetic defect. Nevertheless, our data indicate that NLRP7 is not only a major gene for recurrent moles, but may also be associated with recurrent SAs and a wider spectrum of reproductive wastage. Another interesting finding in this study is that the clinical entity of recurrent SAs, despite its emotional and economic burden to the patients, seems to be, at least in some cases, a milder form of the more severe molar phenotype. This provides an explanation of the lack of Mendelian inheritance of recurrent SAs in families and consequently the lack of success in identifying genes responsible for this clinical entity.
Our cytokine data for patients with A481T with and without other rare NSVs show that these patients, regardless of their reproductive outcomes, with or without HMs, have impaired IL1B and TNF secretion similar to that seen in patients with NLRP7 mutations. These data demonstrate that rare NSVs in NLRP7 have functional consequences and support further their role in conferring susceptibility for recurrent reproductive wastage.
To further investigate the functional consequences of NLRP7 mutations and rare NSVs, we wanted to know what happened in the other term pregnancies of these patients. Histopathological examination of eight placentas from four patients revealed chorioamnionitis, chorangiosis and necrosis, all of which reflect the known functions of NLRP genes, inflammation and apoptosis, and are known factors associated with gynaecological morbidities and perinatal mortalities. Also, among our patients with NLRP7 mutations, six had had seven stillbirths. To investigate the causes of these stillbirths, we examined the histopathology of available placentas and looked for mosaicism and aneuploidies. The absence of identified mosaicism and aneuploides in two studied placentas does not exclude their presence in other regions of these placentas, but the apparent cause of these stillbirths is the various placental abnormalities. The most severe identified abnormality in the two placentas of patient 428 with C399Y was chorioaminionitis, which is a well-known cause of stillbirth and perinatal mortality. This suggests that normal NLRP7 has other roles in late gestation (22–40 weeks), probably in downregulating normal physiological, or pathogen-induced, inflammation, which seems to be severely or mildly impaired by mutations and rare NSVs. Also, the impaired cytokine secretion and consequent inflammatory response of PBMCs from patients with NLRP7 mutations and variants may favour ascension of lower reproductive tract microorganisms and contribute to the preterm rupture of membranes seen in some patients and the occurrence of stillbirths. Taken together, our data indicate that patients with NLRP7 mutations and variants are at higher risk of pregnancy complications and perinatal mortality, an understudied area of medicine affecting a large number of pregnancies. Our data are in agreement with a previous study showing an increased incidence of gynaecological morbidity and perinatal mortality in women with recurrent SAs.13 In addition to the psychological and emotional burden on couples, perinatal mortality constitutes an important economic burden on health systems. Therefore pinpointing its association with NLRP7 mutations and variants will open new avenues of research to better dissect the exact role of NLRP7 in these conditions and help to identify other genes and mechanisms underlying perinatal mortality.
Acknowledgments
We thank the patients, their families and control subjects for their cooperation. We thank Dr Kevin Watters and Nancy Impala at the Department of Pathology of the McGill University Health Centre, the Departments of Pathology of the following hospitals, Sacré-Coeur, Södersjukhuset, Cite de la Santé, Ste Mary, Ste Eustache, Hotel Dieu d'Arthabaska, Anna Laberge, Charles Le Moyne, and Dr Kristofer Delfani for providing paraffin blocks. We also thank Dr W Foulkes for his critical comments on the manuscript, and Dr Martin Laberge for providing clinical information.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
HM Collaborative Group Seang Lin Tan (Montréal, Canada), Alice Benjamin (Montréal, Canada), Lucy Gilbert (Montréal, Canada), Francois Golfier (Montpellier, France), Magali Breguet (Montréal, Canada), Radhika Srinivasan (Chandigarh, India), Adnan El-Hassan (Amman, Jordan), Feodora Stipoljev (Zagreb, Croatia), Srinivasan Krishnamurty (Montréal, Canada), Muhieddine Seoud (Beirut, Lebanon), Ghazi Zaatari (Beirut, Lebanon), Deborah Cohen (Montréal, Canada), Isabelle Girard (Montréal, Canada), R J McKinlay Gardner (Auckland, NewZealand).
Funding RS is supported by a Chercheur Boursier Salary Award, Senior from the FRSQ. CM was supported by a trainee award from the McGill Centre for the Study of Reproduction. RDRB was supported by a CREATE Scholarship from the ‘Reseau Quebecois en Reproduction’. This work is supported by the CIHR (grant number MOP 86546) and the French Ministry of Health to IT.
Competing interests None.
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the McGill Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.