Article Text

Abstract
Introduction We report a 34-year-old Japanese female with a Silver-Russell syndrome (SRS)-like phenotype and a mosaic Turner syndrome karyotype (45,X/46,XX).
Methods/Results Molecular studies including methylation analysis of 17 differentially methylated regions (DMRs) on the autosomes and the XIST-DMR on the X chromosome and genome-wide microsatellite analysis for 96 autosomal loci and 30 X chromosomal loci revealed that the 46,XX cell lineage was accompanied by maternal uniparental isodisomy for all chromosomes (upid(AC)mat), whereas the 45,X cell lineage was associated with biparentally derived autosomes and a maternally derived X chromosome. The frequency of the 46,XX upid(AC)mat cells was calculated as 84% in leukocytes, 56% in salivary cells, and 18% in buccal epithelial cells.
Discussion The results imply that a parthenogenetic activation took place around the time of fertilisation of a sperm missing a sex chromosome, resulting in the generation of the upid(AC)mat 46,XX cell lineage by endoreplication of one blastomere containing a female pronucleus and the 45,X cell lineage by union of male and female pronuclei. It is likely that the extent of overall (epi)genetic aberrations exceeded the threshold level for the development of SRS phenotype, but not for the occurrence of other imprinting disorders or recessive Mendelian disorders.
- Parthenogenesis
- genomic imprinting
- Silver–Russell syndrome
- Turner syndrome
- uniparental disomy
- clinical genetics
- molecular genetics
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Parthenogenesis
- genomic imprinting
- Silver–Russell syndrome
- Turner syndrome
- uniparental disomy
- clinical genetics
- molecular genetics
Although a mammal with maternal uniparental disomy for all chromosomes (upd(AC)mat) is incompatible with life because of genomic imprinting,1 a mammal with a upd(AC)mat cell lineage could be viable in the presence of a co-existing normal cell lineage. In the human, Strain et al2 have reported 46,XX peripheral blood cells with maternal uniparental isodisomy for all chromosomes (upid(AC)mat) in a 1.2-year-old phenotypically male patient with aggressive behaviour, hemifacial hypoplasia and normal birth weight. Because of the 46,XX disorders of sex development, detailed molecular studies were performed, revealing the presence of a normal 46,XY cell lineage in a vast majority of skin fibroblasts and a upid(AC)mat 46,XX cell lineage in nearly all blood cells. In addition, although the data are insufficient to draw a definitive conclusion, Horike et al3 have also identified 46,XX peripheral blood cells with possible upd(AC)mat in a phenotypically male patient through methylation analyses for plural differentially methylated regions (DMRs) in 11 patients with Silver–Russell syndrome (SRS)-like phenotype. This patient was found to have a normal 46,XY cell lineage and a triploid 69,XXY cell lineage in skin fibroblasts.
However, such patients with a upd(AC)mat cell lineage remain extremely rare, and there is no report describing a human with such a cell lineage in the absence of a normal cell lineage. Here, we report a female patient with a upid(AC)mat 46,XX cell lineage and a non-upd 45,X cell lineage who was identified through genetic screenings of 103 patients with SRS-like phenotype.
Materials and methods
Case report
This Japanese female patient was conceived naturally and born at 40 weeks of gestation by a normal vaginal delivery. At birth, her length was 44.0 cm (−3.1 SD), her weight 2.1 kg (−2.9 SD) and her occipitofrontal head circumference (OFC) 30.5 cm (−2.3 SD). The parents and the younger brother were clinically normal (the father died from a traffic accident).
At 2 years of age, she was referred to us because of growth failure. Her height was 77.7 cm (−2.5 SD), her weight 8.45 kg (−2.6 SD) and her OFC 43.5 cm (−2.5 SD). Physical examination revealed several SRS-like somatic features such as triangular face, right hemihypoplasia and bilateral fifth finger clinodactyly. She also had developmental retardation, with a developmental quotient of 56. Endocrine studies for short stature were normal as were radiological studies. Cytogenetic analysis using lymphocytes indicated a low-grade mosaic Turner syndrome (TS) karyotype, 45,X[3]/46,XX[47]. Thus, a screening of TS phenotype4 was performed, detecting horseshoe kidney but no body surface features or cardiovascular lesion. Chromosome analysis was repeated at 6 and 32 years of age using lymphocytes, revealing a 45,X[8]/46,XX[92] karyotype and a 45,X[12]/46,XX[88] karyotype, respectively. On the last examination at 34 years of age, her height was 125.0 cm (−6.2 SD), her weight 37.5 kg (−2.0 SD) and her OFC 51.2 cm (−2.8 SD). She was engaged in a simple work and was able to get on her daily life for herself.
Sample preparation
This study was approved by the Institutional Review Board Committees at National Center for Child health and Development. After obtaining written informed consent, genomic DNA was extracted from leukocytes of the patient, the mother and the brother and from salivary cells, which comprise ∼40% of buccal epithelial cells and ∼60% of leukocytes,5 of the patient. Lymphocyte metaphase spreads and leukocyte RNA were also obtained from the patient. Leukocytes of healthy adults and patients with imprinting disorders were utilised for controls.
Primers and probes
The primers utilised in this study are summarised in supplementary methods and supplementary tables 1–3.
DMR analyses
We first performed bio-combined bisulfite restriction analysis (COBRA)6 and bisulfite sequencing of the H19-DMR (A) on chromosome 11p15.5 by the previously described methods7 and methylation-sensitive PCR analysis of the MEST-DMR (A) on chromosome 7q32.2 by the previously described methods8 with minor modifications (the methylated and unmethylated allele-specific primers were designed to yield PCR products of different sizes, and the PCR products were visualised on the 2100 Bioanalyzer (Agilent, Santa Clara, California, USA)). This was because hypomethylation (epimutation) of the normally methylated H19-DMR of paternal origin and maternal uniparental disomy 7 are known to account for 35–65% and 5–10% of SRS patients, respectively.9 10 In addition, fluorescence in situ hybridisation (FISH) analysis was performed with a ∼84-kb RP5-998N23 probe containing the H19-DMR (BACPAC Resources Center, Oakland, California, USA). We also examined multiple other DMRs by bio-COBRA. The ratio of methylated clones (the methylation index) was calculated using peak heights of digested and undigested fragments on the 2100 Bioanalyzer using 2100 expert software.
Genome-wide microsatellite analysis
Microsatellite analysis was performed for 96 autosomal loci and 30 X chromosomal loci. The segment encompassing each locus was PCR-amplified, and the PCR product size was determined on the ABI PRISM 310 autosequencer using GeneScan software (Applied Biosystems, Foster City, California, USA).
PCR analysis for Y chromosomal loci
Standard PCR was performed for six Y chromosomal loci. The PCR products were electrophoresed using the 2100 Bioanalyzer.
Expression analysis
Quantitative real-time reverse transcriptase PCR analysis was performed for three paternally expressed genes (IGF2, SNRPN and ZAC1) and four maternally expressed genes (H19, MEG3, PHLDA2 and CDKN1C) that are known to be variably (usually weakly) expressed in leukocytes (UniGene, http://www.ncbi.nlm.nih.gov/sites/entrez?db=unigene), using an ABI Prism 7000 Sequence Detection System (Applied Biosystems). TBP and GAPDH were utilised as internal controls.
Results
DMR analyses
In leukocytes, the bio-COBRA indicated severely hypomethylated H19-DMR, and bisulfite sequencing combined with rs2251375 SNP typing for 30 clones revealed maternal origin of 29 hypomethylated clones and non-maternal (paternal) origin of a single methylated clone in this patient (figure 1A). Thus, the marked hypomethylation of the H19-DMR was caused by predominance of maternally derived clones rather than hypomethylation of the H19-DMR of paternal origin. FISH analysis for 100 lymphocyte metaphase spreads excluded an apparent deletion of the paternally derived H19-DMR or duplication of the maternally derived H19-DMR (Supplementary figure 1). Methylation-sensitive PCR amplification for the MEST-DMR delineated a major peak for the methylated allele and a minor peak for the unmethylated allele (figure 1B). This also indicated the predominance of maternally derived clones and the co-existence of a minor portion of paternally derived clones. Furthermore, autosomal DMRs invariably exhibited markedly abnormal methylation patterns consistent with predominance of maternally inherited DMRs, whereas the methylation index of the XIST-DMR on the X chromosome remained within the female reference range (figure 1C). The abnormal methylation patterns were less obvious in salivary cells (thus, in buccal epithelial cells) than in leukocytes, except for the methylation index for the XIST-DMR that mildly exceeded the female reference range (figure 1A–C).
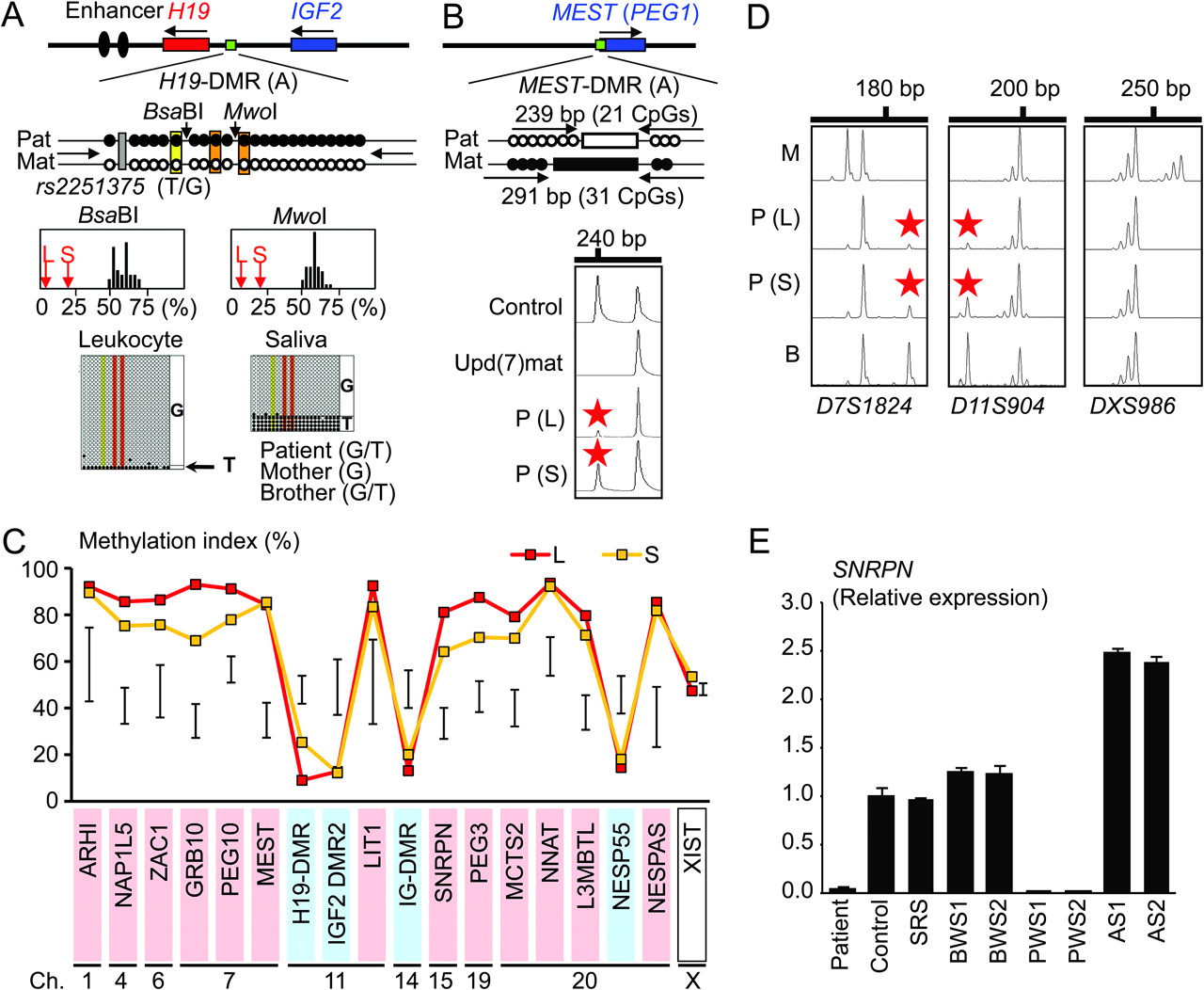
Representative molecular results. Pat, paternally derived allele; Mat, maternally derived allele; P, patient; M, mother; B, brother; L, leukocytes; and S, salivary cells. Filled and open circles in A and B represent methylated and unmethylated cytosine residues at the CpG dinucleotides, respectively. A. Methylation patterns of the H19-DMR (A) harbouring 23 CpG dinucleotides and the T/G SNP (rs2251375) (a grey box). The PCR products are digested with BsaBI when the cytosine at the sixth CpG dinucleotide (highlighted in yellow) is methylated and with MwoI when the two cytosines at the ninth and the 11th CpG dinucleotides (highlighted in orange) are methylated. For the bio-COBRA data, the black histograms represent the distribution of methylation indices (%) in 50 control participants, and L and S denote the methylation indices for leukocytes and salivary cells of this patient, respectively. For the bisulfite sequencing data, each line indicates a single clone. B. Methylated and unmethylated allele-specific PCR analysis for the MEST-DMR (A). In a control participant, the PCR products for methylated and unmethylated alleles are delineated, and the unequal amplification is consistent with a short product being more easily amplified than a long product. In a previously reported patient with upd(7)mat,8 the methylated allele only is amplified. In this patient, major peaks for the methylated allele and minor peaks for the unmethylated allele (red asterisks) are detected. C. Methylation patterns for the 18 DMRs examined. The DMRs highlighted in blue and pink are methylated after paternal and maternal transmissions, respectively. The black vertical bars indicate the reference data (maximum–minimum) in 20 normal control participants, using leukocyte genomic DNA (for the XIST-DMR, 16 female data are shown). D. Representative microsatellite analysis. Minor peaks (red asterisks) have been identified for D7S1824 and D11S904 but not for DXS986 of the patient. Since the peaks for D7S1824 and D11S904 are absent in the mother and clearly present in the brother, they are assessed to be of paternal origin. E. Relative expression level (mean ± SD) of SNRPN on chromosome 15. The data have been normalised against TBP. SRS, an SRS patient with an epimutation (hypomethylation) of the H19-DMR; BWS1, a BWS patient with an epimutation (hypermethylation) of the H19-DMR; BWS2, a BWS patient with upd(11)pat; PWS1, a PWS patient with upd(15)mat; PWS2, a PWS patient with an epimutation (hypermethylation) of the SNRPN-DMR; AS1, an Angelman syndrome (AS) patient with upd(15)pat; and AS2, an AS patient with an epimutation (hypomethylation) of the SNRPN-DMR.
Microsatellite analysis
Major peaks consistent with maternal uniparental isodisomy and minor peaks of non-maternal (paternal) origin were identified for at least one locus on each autosome, with the minor peaks of non-maternal origin being more obvious in salivary cells than in leukocytes (figure 1D and supplementary table 4). Furthermore, the frequency of the upid(AC)mat cells was calculated as 84% in leukocytes, 56% in salivary cells and 18% in epithelial buccal cells, using the area under curves for the maternally and the non-maternally inherited peaks (supplementary note). Such minor peaks of non-maternal origin were not detected for all the 30 X chromosomal loci examined.
PCR analysis for Y chromosomal loci
PCR amplification failed to detect any trace of Y chromosome-specific bands in leukocytes and salivary cells (Supplementary figure 2).
Expression analysis
Expression analysis using control leukocytes indicated that, of the seven examined genes, SNRPN expression alone was strong enough to allow for a precise assessment (Supplementary figure 3). SNRPN expression was extremely low in this patient (figure 1E).
Discussion
These results imply that this patient had a upid(AC)mat 46,XX cell lineage and a non-upd 45,X cell lineage. Indeed, methylation patterns of the XIST-DMR is explained by assuming that the two X chromosomes in the upid(AC)mat cells undergo random X-inactivation and that 45,X cells with the methylated XIST-DMR on a single active X chromosome11 are relatively prevalent in buccal epithelial cells. Furthermore, lack of non-maternally derived minor peaks for microsatellite loci on the X chromosome is explained by assuming that the two X chromosomes in the upid(AC)mat cells and the single X chromosome in the 45,X cells are derived from a common X chromosome of maternal origin, with no paternally derived sex chromosome. It is likely, therefore, that a parthenogenetic activation took place around the time of fertilisation of a sperm missing a sex chromosome, resulting in the generation of the 46,XX cell lineage with upid(AC)mat by endoreplication (the replication of DNA without the subsequent completion of mitosis) of one blastomere containing a female pronucleus and the 45,X cell lineage with biparentally derived autosomes and a maternally derived X chromosome by union of male and female pronuclei (figure 2), although it is also possible that a paternally derived sex chromosome was present in the sperm but was lost from the normal cell lineage at the very early developmental stage. Hence, in a strict sense, this patient is neither a chimera resulting from the fusion of two different zygotes nor a mosaic caused by a mitotic error of a single zygote. In this regard, a triploid cell stage is assumed in the generation of a upid(AC)mat cell lineage, and such triploid cells may have been detected in skin fibroblasts of the patient reported by Horike et al.3

{kind=link}
{kind=link}
Schematic representation of the generation of the upid(AC)mat 46,XX cell lineage and the non-upd 45,X cell lineage. Polar bodies are not shown. PA, parthenogenetic activation; and E, endoreplication of one blastomere containing a female pronucleus.
The upid(AC)mat cells accounted for the majority of leukocytes even in adulthood of this patient, despite global negative selective pressure.12 13 This phenomenon, though intriguing, would not be unexpected in human studies because leukocytes are usually utilised for genetic analyses. Rather, if the upid(AC)mat cells were barely present in leukocytes, they would not have been detected. It is likely, therefore, that upid(AC)mat cells have occupied a relatively large portion of the definitive haematopoietic tissues primarily as a stochastic event. Furthermore, parthenogenetic chimera mouse studies have revealed that parthenogenetic cells are found at a relatively high frequency in some tissues/organs including blood and are barely identified in other tissues/organs such as skeletal muscle and liver.13 Such a possible tissue-specific selection in favour of the preservation of parthenogenetic cells in the definitive haematopoietic tissues may also be relevant to the predominance of the upid(AC)mat cells in leukocytes. In addition, a reduced growth potential of 45,X cells14 may also have contributed to the skewed ratio of the two cell lineages.
Clinical features of this patient would be determined by several factors. They include: (1) the ratio of two cell lineages in various tissues/organs, (2) the number of imprinted regions or DMRs relevant to the development of specific imprinting disorders (eg, plural regions/DMRs on chromosomes 7 and 11 for SRS9 10 and a single region/DMR on chromosome 15 for Prader–Willi syndrome (PWS)),15 (3) the degree of clinical effects of dysregulated imprinted regions/DMRs (an (epi)dominant effect has been assumed for the 11p15.5 imprinted regions including the IGF2–H19 domain on the basis of SRS or Beckwith–Wiedemann syndrome (BWS) phenotype in patients with multilocus hypomethylation16 and BWS-like phenotype in patients with a upid(AC)pat cell lineage,17 a mirror image of a upid(AC)mat cell lineage), (4) expression levels of imprinted genes in upid(AC)mat cells (although SNRPN expression of this patient was consistent with upid(AC)mat cells being predominant in leukocytes, complicated expression patterns have been identified for several imprinted genes in androgenetic and parthenogenetic fetal mice, probably because of perturbed cis- and trans-acting regulatory mechanisms)18 and (5) unmasking of possible maternally inherited recessive mutation(s) in upid(AC)mat cells.19 Collectively, it appears that the extent of overall (epi)genetic aberrations exceeded the threshold level for the development of SRS phenotype and horseshoe kidney characteristic of TS4 but remained below the threshold level for the occurrence of other imprinting disorders or recessive Mendelian disorders.
In summary, we identified a upid(AC)mat 46,XX cell lineage in a woman with an SRS-like phenotype and a 45,X cell lineage accompanied by autosomal haploid sets of biparental origin. This report will facilitate further identification of patients with a upid(AC)mat cell lineage and better clarification of the clinical phenotypes in such patients.
Acknowledgments
We thank the patient and her family members for their participation in this study. We also thank Dr. Toshiro Nagai for providing us with blood samples of patients with Prader–Willi syndrome.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
Funding This work was supported by grants from the Ministry of Health, Labor, and Welfare and from the Ministry of Education, Science, Sports and Culture.
Competing interests None.
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the Institutional Review Board Committees at National Center for Child health and Development.
Provenance and peer review Not commissioned; externally peer reviewed.