Article Text

Abstract
Background Radiotherapy-induced DNA double-strand breaks (DSBs) are critical cytotoxic lesions. Inherited defects in DNA DSB repair pathways lead to hypersensitivity to ionising radiation, immunodeficiency and increased cancer incidence. A patient with xeroderma pigmentosum complementation group C, with a scalp angiosarcoma, exhibited dramatic clinical radiosensitivity following radiotherapy, resulting in death. A fibroblast cell line from non-affected skin (XP14BRneo17) was hypersensitive to ionising radiation and defective in DNA DSB repair.
Aim To determine the genetic defect causing cellular radiation hypersensitivity in XP14BRneo17 cells.
Methods Functional genetic complementation whereby copies of human chromosomes containing genes involved in DNA DSB repair (chromosomes 2, 5, 8 10, 13 and 22) were individually transferred to XP14BRneo17 cells in an attempt to correct the radiation hypersensitivity. Clonogenic survival assays and γ-H2AX immunofluorescence were conducted to measure radiation sensitivity and repair of DNA DSBs. DNA sequencing of defective DNA repair genes was performed.
Results Transfer of chromosome 8 (location of DNA-PKcs gene) and transfection of a mammalian expression construct containing the DNA-PKcs cDNA restored normal ionising radiation sensitivity and repair of DNA DSBs in XP14BRneo17 cells. DNA sequencing of the DNA-PKcs coding region revealed a 249-bp deletion (between base pairs 3656 and 3904) encompassing exon 31 of the gene.
Conclusion We provide evidence of a novel splice variant of the DNA-PKcs gene associated with radiosensitivity in a patient with xeroderma pigmentosum and report the first double mutant in distinct DNA repair pathways being consistent with viability.
- Functional complementation
- radiosensitivity
- DNA repair
- DNA-PKcs gene
- pre-mRNA splicing
- genetics
- molecular genetics
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Functional complementation
- radiosensitivity
- DNA repair
- DNA-PKcs gene
- pre-mRNA splicing
- genetics
- molecular genetics
Introduction
Anticancer radiotherapy (RT) protocols exert cytotoxicity by inducing DNA damage, predominantly DNA double-strand breaks (DSBs). Human cells possess enzymatic DNA repair systems which monitor and repair such damage, and the efficacy of DNA repair has a significant impact on the success of RT1. In mammalian cells, two distinct biochemical pathways are responsible for repairing DSBs, homologous recombination and non-homologous end joining (NHEJ).2 NHEJ is the predominant repair pathway whereby the DNA-dependent protein kinase heterodimer containing the Ku80–Ku70 complex binds to DNA ends and recruits DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and its substrate, the Artemis nuclease which prepares DNA ends for ligation. Ligation is mediated by a protein complex containing DNA ligase IV stabilised and stimulated by XRCC4 and Cernunnos-XLF.3 Animal models show that defects in any component of NHEJ can lead to hypersensitivity to ionising radiation (IR), genome instability, immunodeficiency and cancer.4 In humans, defects in the specific genetic components of NHEJ also result in predisposition to lymphoma and leukaemia or to extreme radiosensitivity during RT.5 6
Xeroderma pigmentosum (XP) is a rare autosomal recessive disease characterised by sensitivity to sunlight and susceptibility to skin cancers.7 There are seven XP complementation groups (A–G) which define defects in nucleotide excision repair (NER), responsible for excising UV-induced DNA photodamage.8 Although cells from XP patients are not typically sensitive to IR, cellular radiosensitivity has been reported in fibroblasts from an XPG patient and in an XPC patient.9 10 The XPC patient with a truncation in the C-terminus of the XPC protein presented with an angiosarcoma of the scalp, which was treated by surgical excision and RT.11 Following 38 Gy in 19 fractions with 6 MeV electrons, a severe scalp desquamation occurred associated with necrosis of the underlying bone, intracranial oedema and death.
A primary fibroblast cell line (XP14BR) and an SV40-transformed derivative cell line (XP14BRneo17) from this patient were hypersensitive to the lethal effects of IR and UV, consistent with the clinical phenotype.11 Transfection with the XPC cDNA restored a normal UV cytotoxic response, but IR sensitivity was unchanged. XP14BR fibroblasts were distinguishable from radiosensitive ataxia telangiectasia cells by virtue of a normal inhibition of DNA synthesis after IR exposure. Repair of radiation-induced DNA DSBs in XP14BR cells was significantly reduced compared with normal fibroblast cell lines.11 XP14BR fibroblasts were not sensitive to the lethal effects of the DNA interstrand cross-linking agents, mitomycin C or nitrogen mustard, indicating that these cells were not defective in homologous recombination, which plays an important role in regulating interstrand cross-linking cytotoxicity.12
In this paper we present evidence, from microcell-mediated chromosome transfer (MMCT), expression plasmid transfer and sequence analysis, that NHEJ was defective in this patient as a result of a defect in the DNA-PKcs gene. The molecular basis of this IR hypersensitivity is deletion of exon 31 of the DNA-PKcs gene causing an in-frame deletion of 249 bp with a predicted 82-amino acid deletion from the protein.
This is the second reported case of a defective DNA-PKcs gene in human cells. It is also the first reported case in which two distinct DNA repair pathways (NER and NHEJ) are defective in a single individual. These observations indicate how undiagnosed defects in DNA repair pathways leave patients at risk of life-threatening toxicity during RT and imply the need for a simple pretreatment screening protocol to identify individuals at risk.
Methods
Cell culture
The primary fibroblast cell lines XP14BR, MRC5, NB1 and 1BR.3 and culture conditions are described in detail elsewhere.10 13 14 The SV40-immortalised fibroblast cell lines XP14BRneo17, 1BR.3-G, MRC5-SV1 and GMO637 and culture conditions are all described elsewhere.11 Culture of the MO59J human glioblastoma cell line used for western blot analysis has been previously described.15
Clonogenic cell survival assays
Clonogenic cell survival assays after exposure to γ-radiation (from a 60Cobalt source) were conducted as previously described.11 13
Microcell-mediated chromosome transfer
MMCT was performed as described elsewhere.16 Briefly, A9–monochromosome donor hybrids were exposed to 75–100 ng/ml colcemid (Sigma, Poole, Dorset, UK) for 48 h to induce metaphase arrest and micronucleation. After a 5 min incubation in serum-free medium containing 10 μg/ml cytochalasin B (Sigma), micronuclei were harvested by centrifugation at 4500 rpm for 15 min at room temperature and purified by successive filtration through 8, 5 and 3 μm polycarbonate filters (Whatman, Kent, UK). Purified micronuclei were fused to recipient cells in the presence of 100 μg/ml phytohaemaglutinin B, 10% dimethyl sulfoxide and poly(ethylene glycol) (Sigma) for 1 min. Monochromosome hybrids of XP14BRneo17 were selected 48 h later by incubating the cells in the presence of 150 μg/ml hygromycin B (Merck Chemicals Ltd, Nottingham, UK). After 3 weeks, hygromycin B-resistant colonies were isolated.
γ-Irradiation prescreen of XP14BRneo17 monochromosome hybrids
To identify chromosomes that functionally correct the IR hypersensitivity of XP14BRneo17 cells, hybrids were exposed to 2 Gy γ-radiation. Five thousand cells were plated, and the number of colonies formed after 3 weeks were expressed as a percentage of colonies formed from non-γ-irradiated cells. Survival of XP14BRneo17 monochromosome hybrids at 2 Gy was compared with that of XP14BRneo17 parental cells and the repair normal cell line 1BR.3-G.
cDNA transfection
The cDNA expression constructs of DNA-PKcs and Ku80 were a gift from Dr David Chen (Department of Radiation Oncology, University of Texas Southwestern Medical Center, Dallas, Texas, USA). The plasmid pCMPD-6 using the cytomegalovirus (CMV) promoter contained a full-length human DNA–PKcs cDNA. The pKM-A1 plasmid (under control of the CMV promoter) contained a full-length DNA–PKcs cDNA with an inactivating missense lysine-to-arginine mutation at amino acid 3752 in the serine/threonine kinase domain of the protein.17 The Ku80 cDNA was provided in the pIREShyg2 plasmid (BD Biosciences, Cowley, Oxford, UK), with gene expression under the control of the CMV promoter.
Transfection of XP14BRneo17 cells with the DNA-PKcs or Ku80 expression plasmids was performed using calcium phosphate coprecipitation.17 The pCMPD-6 and pKM-A1 plasmids with the DNA-PKcs cDNA were cotransfected with the pPur plasmid (Clontech, Basingstoke, Hants, UK), conferring resistance to the antibiotic, puromycin. Then 15 μg of each plasmid was added to the cells for 24 h, after which cells were selected in 100 ng/ml puromycin for 4 weeks. For pIREShyg2-Ku80 plasmid transfections, cells were selected in 150 units/ml hygromycin B.
Detection of γ-H2AX foci: immunofluorescence assay
Nuclear γ-H2AX foci after 2 Gy irradiation (at 0, 1, 5, 7, 9 and 24 h) were detected as described previously.18 Monoclonal antibody to phosphoserine139 γ-H2AX (Millipore (U.K.) Limited, Croxley Green Business Park, Watford, UK) at 1:10 000 dilution was added for 1 h at room temperature. Alexa Fluor 488 goat anti-mouse IgG antibody diluted 1:1000 was added to the cells for 1 h (Molecular Probes, Paisley, UK), after which they were counterstained with 2 μg/ml propidium iodide in phosphate-buffered saline and mounted in Vectashield (Vector Laboratories, Peterborough, UK). Analysis was performed by counting the number of foci in a minimum of 100 nuclei of each cell line per time point using a fluorescence microscope. Results are expressed as the percentage of γ-H2AX-positive cells containing five or more γ-H2AX foci, as untreated cells typically exhibited 0–2 foci per/cell.
Western blot analysis
Protein extracts of XP14BR, MRC5, 1BR.3 primary human fibroblast and MO59J human glioblastoma cell line, derived from 2×105 cells, were separated by sodium dodecyl sulfate/polyacrylamide gel electrophoresis using a 4–15% gradient gel (Bio-Rad Laboratories Ltd, Hemel Hempstead, Herts, UK). Proteins of interest were detected by western blotting using a mouse anti-(human DNA-PKcs) monoclonal antibody (Lab Vision, Freemont, California, USA) or human β-actin as a loading control (Abcam PLC, Cambridge, UK), and proteins were visualised by chemiluminescence.
DNA sequence analysis of the DNA-PKcs coding region
Total RNA was isolated from primary XP14BR cells using TriZol reagent (Sigma). cDNA was synthesised from total RNA using random primers and Superscript III reverse transcriptase (InVitrogen). The DNA-PKcs cDNA sequence from XP14BR encompassing 12 291 bp was constructed in a contig of 17 fragments between 800 and 1000 bp in size using gene-specific primers by PCR. Each amplified fragment overlapped by ∼100 bp. cDNA fragments were cloned into the pCR-4-Topo sequencing vector (InVitrogen). Four individual clones of each fragment of DNA-PKcs from XP14BR cells were subjected to DNA sequence analysis on both the coding and non-coding strand.
Results
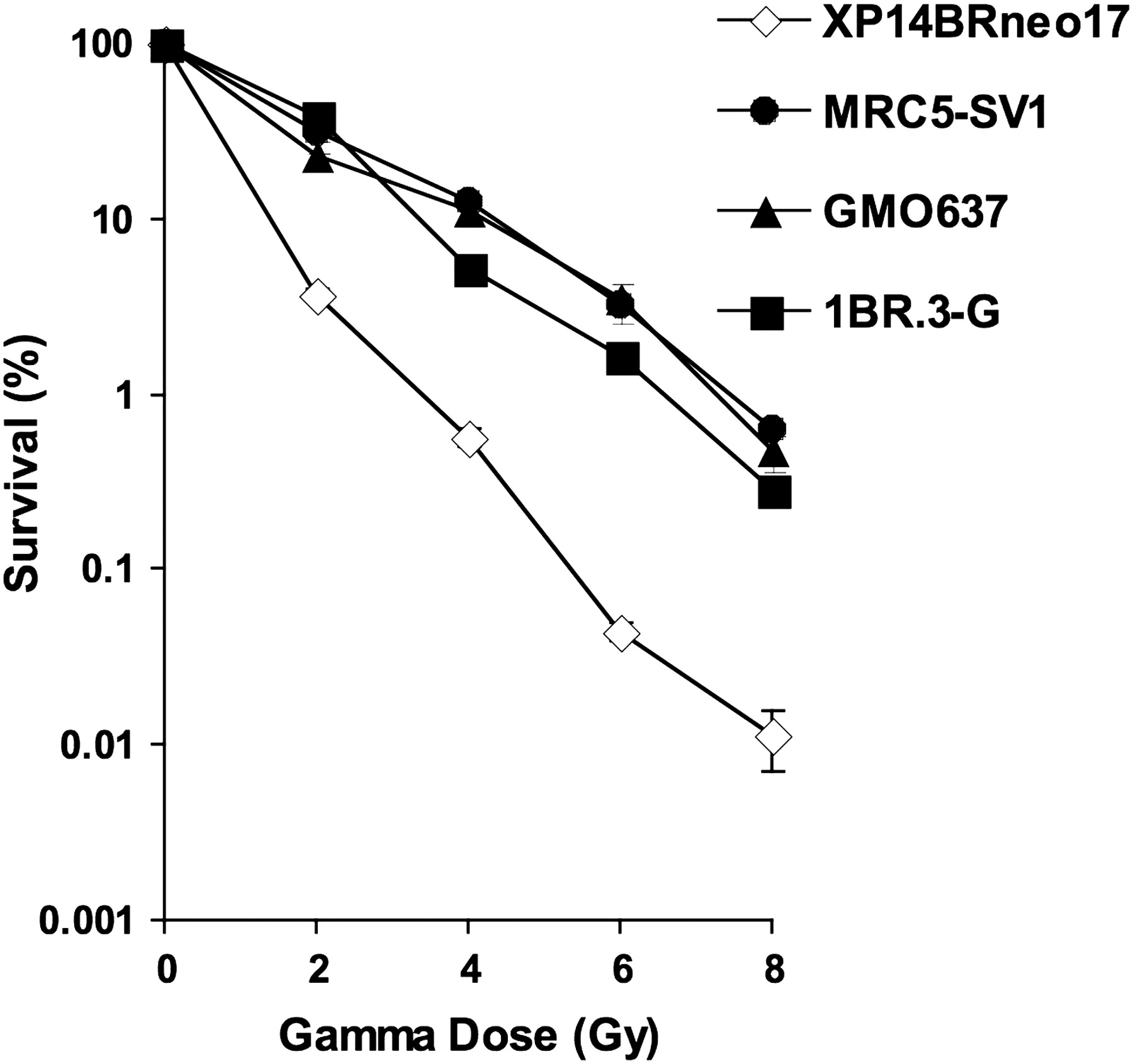
The dose required to reduce clonogenic cell survival to 10% in immortalised DNA repair normal cell lines, 1BR.3-G, MRC5-SV1 and GM0637, was 3.5, 4.8 and 4.8 Gy, respectively. For the XP14BRneo17 cell line, 1.4 Gy reduced the clonogenic survival to 10% (figure 1). Hence XP14BRneo17 cells were 2.5–3.4-fold more sensitive to the lethal effects of γ-irradiation than the three normal cell lines tested.
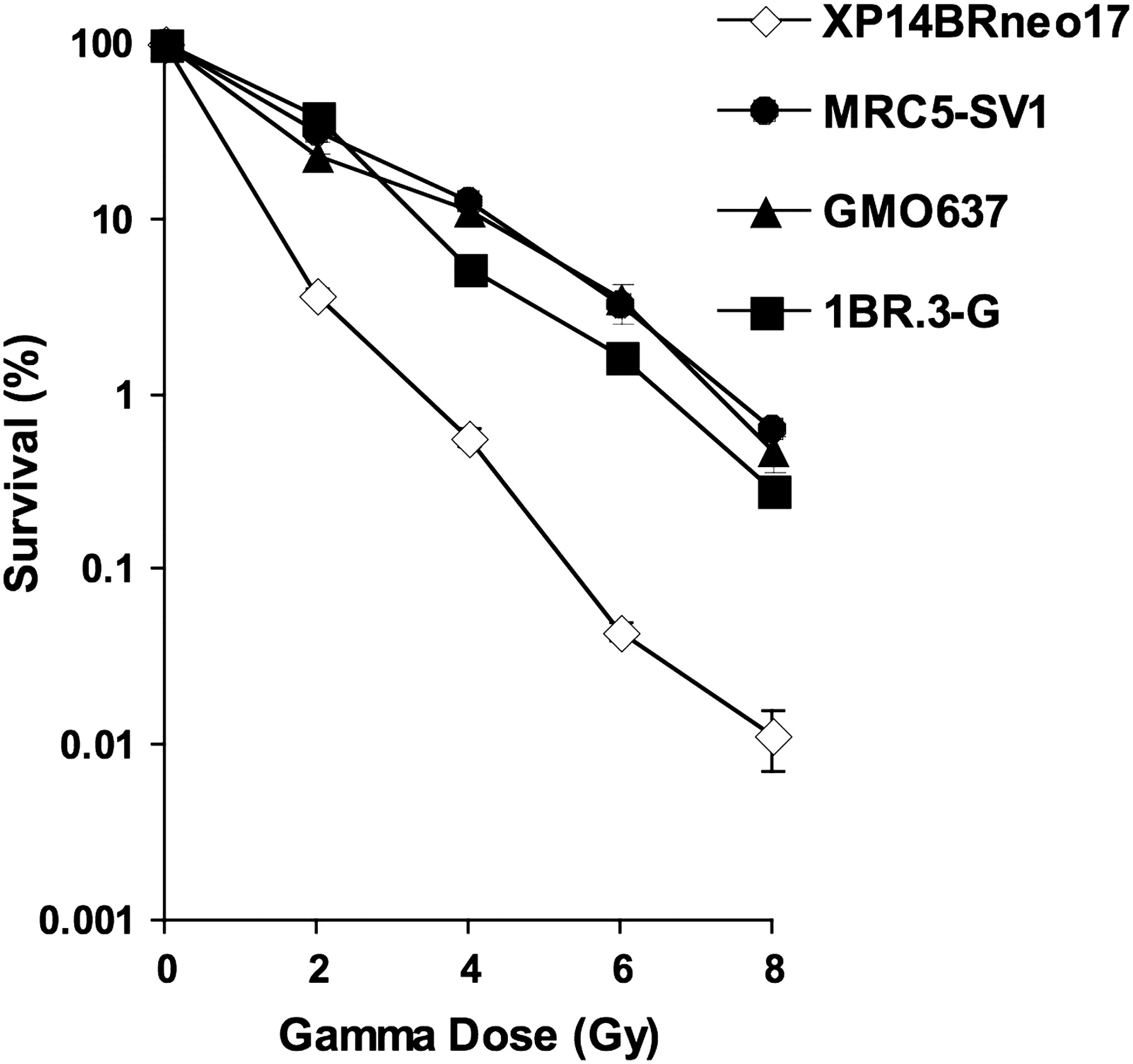
Clonogenic cell survival of XP14BRneo17 compared with MRC5-SV1, GM0637 and 1BR.3-G after exposure to increasing doses of γ-radiation. The error bars represent the SEM response derived from three independent experiments.
Normal copies of six individual human chromosomes harbouring NHEJ genes required for DSB repair were individually transferred to XP14BRneo17 cells (table 1) and screened for radiation sensitivity after exposure to 2 Gy γ-irradiation (figure 2A). In all radiation-sensitive hybrids and in XP14BRneo17 cells, the cell survival was ∼10% or less. Transfer of chromosome 8 in one hybrid (Chr.8 C) resulted in 46% cell survival, in line with the level of cell survival expected in the repair normal cell line 1BR.3-G. That only one hybrid containing chromosome 8 corrected the radiation-sensitivity phenotype is consistent with previous findings in which only a small proportion of hybrids exhibited functional complementation.16 The prolonged colcemid exposure during MMCT to induce micronucleation introduces small intrachromosomal deletions in donor chromosomes, which often encompass key loci, which, in these experiments, may include the large DNA-PKcs genomic sequence (∼200 kb). Complete clonogenic survival assays showing a normal cell survival response of hybrid Chr.8 C similar to the repair normal 1BR.3-G, MRC5-SV1 and GM0637 cells are presented in figure 2B. As chromosome 8 (8q11) contains the DNA-PKcs gene required for NHEJ, this provided evidence that a mutation in DNA-PKcs may account for the clinical and cellular radiosensitivity of this patient.

A) Clonogenic cell survival after exposure to 2 Gy γ-radiation of XP14BRneo17-monochromosome hybrids, parental XP14BRneo17 cells and repair normal 1BR.3-G cells. The hybrid of chromosome 8 functionally correcting the ionising radiation (IR) hypersensitivity of XP14BRneo17 cells is indicated as Chr.8 C. (B) Clonogenic cell survival of XP14BRneo17 compared with MRC5-SV1, GM0637 and 1BR.3-G and hybrids of XP14BRneo17 cells containing an additional human chromosome 8, after exposure to increasing doses of γ-radiation. The chromosome 8 hybrid exhibiting a corrected IR response is referred to as Chr.8 C. The error bars represent the SEM response derived from three independent experiments.
Chromosomes containing genes involved in NHEJ together with the number of hybrids derived from each MMCT experiment
XP14BRneo17 cells were transfected with an expression plasmid containing the DNA-PKcs cDNA (pCMPD-6). As a control, XP14BRneo cells were also transfected with an expression plasmid containing a non-functional mutation in the kinase domain of the DNA-PKcs cDNA (pPKM-A1). Figure 3A shows that two clones of XP14BRneo17 transfected with pCMPD-6 had a normal clonogenic cell survival response. XP14BRneo17 cells transfected with the mutated DNA-PKcs cDNA or the Ku80 cDNA remained radiation sensitive. Restoration of a normal IR response by DNA-PKcs transfection in XP14BRneo 17 cells was associated with efficient DNA DSB repair, measured by the induction and removal of γ-H2AX foci in the nuclei of cells irradiated with 2 Gy γ-radiation. Representative examples of γ-H2AX foci staining are shown in figure 3B. Immediately after γ-irradiation, over 80% of all cells had five or more γ-H2AX foci. Figure 3C shows that the normal level of radiosensitivity exhibited by transfection of XP14BRneo17 cells with the DNA-PKcs cDNA is associated with the removal of γ-H2AX foci similar to the normal fibroblasts MRC5-SV1 and GM0637. After 24 h, 65.5% of XP14BRneo17 cells retained five or more γ-H2AX foci. Also, in XP14BRneo17 cells transfected with the mutated DNA-PKcs or the Ku80 cDNA, 70.11% and 76.20%, respectively, of nuclei scored positive for γ-H2AX foci after 24 h. Two clones of XP14BRneo17 cells transfected with normal DNA-PKcs showed a reduction of positive nuclei to 12.3% and 18.6% after 24 h, which was similar to the normal MRC5-SV1 and GMO636 cell lines.

(A) Clonogenic cell survival of XP14BRneo17 compared with MRC5-SV1, GM0637 and 1BR.3-G and XP14BRneo17 cells after transfection with full-length DNA-PKcs cDNA (pCMPD-6 clones 1 and 2) or DNA-PKcs cDNA with a mutation in the kinase domain (pPKM-A1, clones 1 and 2) and XP14BRneo17 cells transfected with the Ku80 cDNA, after exposure to increasing doses of γ-radiation. The error bars represent the SEM response derived from three independent experiments. (B) A representative image of γ-H2AX foci in the nuclei of XP14BRneo 17 and a clone of XP14BR neo17 transfected with the DNA-PKcs cDNA expression plasmid pCMPD-6 (clone 1) after exposure to 2 Gy γ-irradiation. (C) Induction and repair over a 24 h period of 2 Gy γ-radiation-induced γ-H2AX foci in the nuclei of irradiated cells. MRC5-SV1 and GMO637 are repair normal cell lines. The ionising radiation (IR)-hypersensitive XP14BRneo17 parental cell line is compared with XP14BRneo17 cells transfected with either pCMPD-6 (normal DNA-PKcs) or pPKM-A1 (mutated DNA-PKcs) and pIREShyg2-Ku80 (normal Ku80 cDNA). Error bars represent the SEM of two independent experiments for each cell line.
We then investigated whether the DNA-PKcs protein was produced in the XP14BR cells. DNA-PKcs protein expression levels in XP14BR, MRC-5, 1BR.3 and MO59J cells detected by western blot are shown in figure 4. In XP14BR cells, a protein that comigrates with full-length DNA-PKcs protein is expressed when compared with DNA repair normal MRC-5 and 1BR.3 cells. However, taking into account the mass of the DNA-PKcs protein (470 kDa), it was not possible to detect small in-frame deletions by western blotting. A band could not be detected in the MO59J cells, consistent with a deletion of DNA-PKcs in this repair-defective cell line.15

(A) Western blot analysis of DNA-PKcs protein expression in primary XP14BR compared with repair normal MRC-5 and 1BR.3 human fibroblasts and DNA-PKcs defective MO59J human glioblastoma cell line. (B) As a loading control, human β-actin expression in each cell type was also examined.
To identify the mutation, extensive DNA sequence analysis of 136 cloned fragments (four independent clones of each of the 17 fragments sequenced on the sense and antisense strand) of the cDNA contig of DNA-PKcs of XP14BR revealed a 249 bp in-frame deletion between base pairs 3656 and 3904 encompassing the entire exon 31, resulting in a predicted 82-residue deletion. This was observed in three of the four independent clones sequenced. The location of this deletion within the context of the DNA-PKcs coding sequence is shown in figure 5. To identify what would cause the exon skipping, we sequenced the genomic DNA surrounding exon 31. Sequencing of the surrounding intron/exon junctions, including 250 bp intronic DNA either side of exon 31 in genomic DNA, did not reveal a splicing mutation. To confirm these observations, the cDNA encompassing this region was sequenced from a second independent isolate of RNA from XP14BR cells, and the exon 31 deletion was observed in two of four clones. Sequence analysis of cDNA encompassing exons 30–32 of DNA-PKcs from two normal cell lines (1BR.3 and NB1: four independent cloned fragments from each cell line) revealed no such deletion.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic view of the DNA-PKcs protein. Functional domains, identified from the Pfam database (http://pfam.sanger.ac.uk/), and the location of exon 31 are highlighted. Comparisons with the DNA-PKcs protein sequences (NCBI) from other species from within the exon 31 region are shown in an expanded view. Boxed sequences highlight evolutionarily conserved residues.
Discussion
In this study we report a primary defect in the DNA-PKcs gene in normal (non-tumour) tissue from a human patient. DNA-PKcs is an essential genetic component in the repair of ionising radiation-induced DNA DSB via the NHEJ pathway The DNA-PK complex consists of Ku70/80 heterodimer and a 470 kDa catalytic subunit DNA-PKcs with serine/threonine kinase activity. DNA-PKcs binds to DNA ends in a Ku80-dependent manner, to initially suppress efficient ligation of DNA ends. DNA-PKcs autophosphorylation then liberates the DNA ends of the strand break, allowing ligation by the ligase IV–XRCC4 protein complex. It is speculated that DNA-PKcs acts as a molecular ‘gatekeeper’ preventing premature ligation of DNA ends.19
Animal models have shown that defects in any component of NHEJ (including DNA-PKcs) can lead to hypersensitivity to IR, genome instability, immunodeficiency and cancer.4 Until recently, in humans, only two of these genes, ligase IV and Artemis, have been linked to a predisposition for lymphoma or leukaemia and to extreme radiosensitivity.5 6 The first report that mutations in the DNA-PKcs gene can result in radiosensitivity and severe combined immunodeficiency involved a patient with a missense mutation in the DNA-PKcs gene (L3062R) causing an inability to activate Artemis leading to defective NHEJ.20
To define the molecular defect in DNA-PKcs in XP14BR cells, western blot analysis (figure 4) indicated that a protein indistinguishable in length from the DNA-PKcs protein was detectable when compared with the normal 1BR.3 and MRC5 cells. These data suggested that it was unlikely that a large structural deletion in the DNA-PKcs gene was responsible for the defective function of the protein. DNA sequence analysis of the DNA-PKcs cDNA in XP14BR revealed a 249 bp deletion of the entire exon 31 of the transcript, resulting in a predicted 82-amino acid deletion. In two normal cell lines (1BR.3 and MRC5), exon 31 was retained in the full-length transcript. Analysis of the intron/exon boundaries of exon 31 did not reveal a splicing mutation.
There are two possibilities for why this splicing defect might cause the radiosensitivity in XP14BR cells. Firstly, the patient is heterozygous and therefore haploinsufficient for the DNA-PKcs protein; however, we did not identify any genomic mutation causing the splicing abnormality in the DNA-PKcs coding sequence. Secondly, we have identified a novel splicing isoform of DNA-PKcs (EX31) which is expressed in the XP14BR patient, and the EX31 protein is acting as a dominant-negative mutant.
The functional consequences of the predicted 82-amino acid deletion from the protein are summarised in figure 5. Multiple sequence alignments (http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) and structural analysis (http://www.ics.uci.edu/∼baldig/) indicate that the 82-amino acid sequence encoded by exon 31 could be folded into an α-helix. Conservation of amino acid residues between species indicates that the region may have an important role in maintaining protein structure.
The deletion of an entire exon without an apparent splicing mutation is not normally considered to be an unusual observation. For DNA-PKcs, at least three conserved splice variants, including a full-length transcript (variant I), a transcript that retains an intron between exon 82 and 83 (variant II), and a transcript in which exon 80 is deleted (variant III), are observed.21
Pre-mRNA splicing is an essential step in the expression of almost all eukaryotic genes, and alternative splicing appears to be the mechanism responsible for the generation of up to 75% of the human proteome. There are different modes of alternative splicing; however, the differential inclusion or exclusion of an exon or group of exons, so-called cassette exon, is the most common type of alternative splicing event.22
In summary, we conclude that the deletion of exon 31 of the DNA-PKcs gene in the XP14BR patient represents a novel isoform of the DNA-PKcs gene whose expression is associated with the clinical and cellular IR hypersensitivity. Interestingly, immunodeficiency, often observed with human defects of NHEJ, was not observed in this patient23. We provide only the second reported case of an individual with a primary defect in the DNA-PKcs gene, but the first report of an individual with DNA repair defects in two independent pathways, NHEJ and NER.
Finally, these data show how seemingly ‘mild’ or undiagnosed defects in DNA repair factors, while consistent with viability, can have catastrophic consequences should such an individual require cytotoxic anticancer RT. Simple pretreatment screening protocols such as measuring the induction of repair of nuclear γ-H2AX foci in patient cells,24 to identify individuals at risk, would increase the safety of RT for such patients.
References
Footnotes
Funding The St Bartholomew's and the London NHS Trust Charity.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.